
The Therapeutic Monoclonal Antibody Bamlanivimab Does Not Enhance SARS-CoV-2 Infection by FcR-Mediated Mechanisms

 , , , and add
Show full author list
, , , and add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. In Vitro Methods
2.1.1. Cell Culture
2.1.2. Flow Cytometry
2.1.3. RT-PCR
2.1.4. Surface Plasmon Resonance
2.1.5. C1q ELISA
2.2. In Vitro Cell-Based Assays
2.2.1. Dilution of Test Samples
2.2.2. Preparation of CHO-Spike/hCD20 Target Cells
2.2.3. Preparation of Jurkat FcγRIIIa (V158)-NFAT-Luc Cells
2.2.4. Preparation of Complement
2.2.5. Luminescence Readout
2.3. ADCP Assay
2.3.1. Differentiation of Macrophages from Primary Monocytes
2.3.2. Cell Labeling, Antibody Treatment and Co-Culture, and Data Acquisition
2.3.3. ADCP Assay Data Analysis
2.4. In Vivo Methods
2.4.1. Virus
2.4.2. Animal Challenge
2.4.3. Virus Titration
2.4.4. RNA Isolation from SARS-CoV-2-Infected AGM Samples
2.4.5. Detection of SARS-CoV-2 Load from AGMs
2.4.6. Hematology and Serum Biochemistry
2.4.7. Serum ELISAs for Human IgG Concentrations
2.4.8. Serum Neutralization Assay
2.4.9. Bead-Based Cytokine and Coagulation Immunoassays
2.5. Pathology and Immunohistochemistry
2.5.1. Pathology
2.5.2. Immunohistochemistry
2.5.3. Pancytokeratin
2.6. Statistics
Virology Statistics
3. Results
3.1. Evaluation of In Vitro ADE
3.2. FcγR Binding
3.3. Antibody-Dependent Cell-Mediated Cytotoxicity and Complement-Dependent Cytotoxicity
3.4. Antibody-Dependent Cellular Phagocytosis
3.5. In Vivo Study Design
3.6. Quantification of SARS-CoV-2 Viral Loads in AGMs
3.7. Clinical Pathology and Quantification of Serum Inflammatory Mediators
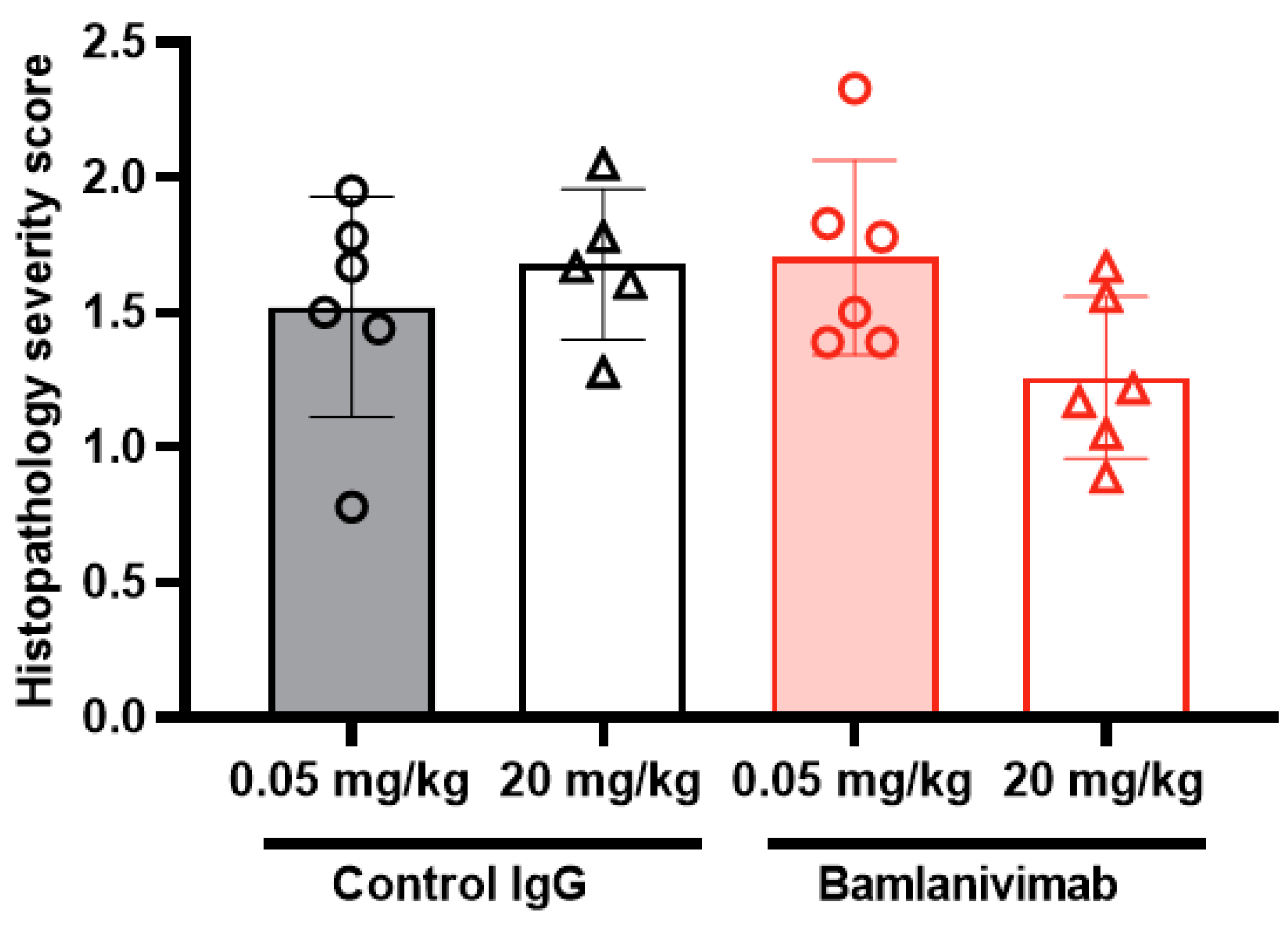
3.8. Histology and Immunohistochemistry
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Jones, B.E.; Brown-Augsburger, P.L.; Corbett, K.S.; Westendorf, K.; Davies, J.; Cujec, T.P.; Wiethoff, C.M.; Blackbourne, J.L.; Heinz, B.A.; Foster, D.; et al. LY-CoV555, a rapidly isolated potent neutralizing antibody, provides protection in a non-human primate model of SARS-CoV-2 infection. bioRxiv 2020. [Google Scholar] [CrossRef]
- Chen, P.; Nirula, A.; Heller, B.; Gottlieb, R.L.; Boscia, J.; Morris, J.; Huhn, G.; Cardona, J.; Mocherla, B.; Stosor, V.; et al. SARS-CoV-2 Neutralizing Antibody LY-CoV555 in Outpatients with COVID-19. N. Engl. J. Med. 2020, 384, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.C.; Adams, A.C.; Hufford, M.M.; de la Torre, I.; Winthrop, K.; Gottlieb, R.L. Neutralizing monoclonal antibodies for treatment of COVID-19. Nat. Rev. Immunol. 2021, 21, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Gajjar, M.R.; Yu, J.; Padte, N.N.; Gettie, A.; Blanchard, J.L.; Russell-Lodrigue, K.; Liao, L.E.; Perelson, A.S.; Huang, Y.; et al. Quantifying the contribution of Fc-mediated effector functions to the antiviral activity of anti–HIV-1 IgG1 antibodies in vivo. Proc. Natl. Acad. Sci. USA 2020, 117, 18002–18009. [Google Scholar] [CrossRef]
- Saphire, E.O.; Schendel, S.L.; Gunn, B.M.; Milligan, J.C.; Alter, G. Antibody-mediated protection against Ebola virus. Nat. Immunol. 2018, 19, 1169–1178. [Google Scholar] [CrossRef]
- Lu, L.L.; Suscovich, T.J.; Fortune, S.M.; Alter, G. Beyond binding: Antibody effector functions in infectious diseases. Nat. Rev. Immunol. 2018, 18, 46–61. [Google Scholar] [CrossRef]
- Bournazos, S.; Gupta, A.; Ravetch, J.V. The role of IgG Fc receptors in antibody-dependent enhancement. Nat. Rev. Immunol. 2020, 20, 633–643. [Google Scholar] [CrossRef]
- Lee, W.S.; Wheatley, A.K.; Kent, S.J.; DeKosky, B.J. Antibody-dependent enhancement and SARS-CoV-2 vaccines and therapies. Nat. Microbiol. 2020, 5, 1185–1191. [Google Scholar] [CrossRef]
- Polack, F.P. Atypical measles and enhanced respiratory syncytial virus disease (ERD) made simple. Pediatr. Res. 2007, 62, 111–115. [Google Scholar] [CrossRef]
- Kuzmina, N.A.; Younan, P.; Gilchuk, P.; Santos, R.I.; Flyak, A.I.; Ilinykh, P.A.; Huang, K.; Lubaki, N.M.; Ramanathan, P.; Crowe, J.E., Jr.; et al. Antibody-dependent enhancement of Ebola virus infection by human antibodies isolated from survivors. Cell Rep. 2018, 24, 1802–1815.e5. [Google Scholar] [CrossRef]
- Katzelnick, L.C.; Gresh, L.; Halloran, M.E.; Mercado, J.C.; Kuan, G.; Gordon, A.; Balmaseda, A.; Harris, E. Antibody-dependent enhancement of severe dengue disease in humans. Science 2017, 358, 929–932. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.T.; Sewatanon, J.; Memoli, M.J.; Wrammert, J.; Bournazos, S.; Bhaumik, S.K.; Pinsky, B.A.; Chokephaibulkit, K.; Onlamoon, N.; Pattanapanyasat, K.; et al. IgG antibodies to dengue enhanced for FcγRIIIA binding determine disease severity. Science 2017, 355, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Halstead, S.; O’rourke, E. Dengue viruses and mononuclear phagocytes. I. Infection enhancement by non-neutralizing antibody. J. Exp. Med. 1977, 146, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Dejnirattisai, W.; Supasa, P.; Wongwiwat, W.; Rouvinski, A.; Barba-Spaeth, G.; Duangchinda, T.; Sakuntabhai, A.; Cao-Lormeau, V.-M.; Malasit, P.; Rey, F.A.; et al. Dengue virus sero-cross-reactivity drives antibody-dependent enhancement of infection with zika virus. Nat. Immunol. 2016, 17, 1102–1109. [Google Scholar] [CrossRef]
- Sánchez-Zuno, G.A.; Matuz-Flores, M.G.; González-Estevez, G.; Nicoletti, F.; Turrubiates-Hernández, F.J.; Mangano, K.; Muñoz-Valle, J.F. A review: Antibody-dependent enhancement in COVID-19: The not so friendly side of antibodies. Int. J. Immunopathol. Pharmacol. 2021, 35, 20587384211050199. [Google Scholar] [CrossRef] [PubMed]
- Yip, M.; Leung, H.; Li, P.; Cheung, C.; Dutry, I.; Li, D.; Daeron, M.; Bruzzone, R.; Peiris, J.S.; Jaume, M. Antibody-dependent enhancement of SARS coronavirus infection and its role in the pathogenesis of SARS. Hong Kong Med. J. 2016, 22, 25–31. [Google Scholar] [CrossRef]
- Yip, M.S.; Leung, N.H.L.; Cheung, C.Y.; Li, P.H.; Lee, H.H.Y.; Daëron, M.; Peiris, J.S.M.; Bruzzone, R.; Jaume, M. Antibody-dependent infection of human macrophages by severe acute respiratory syndrome coronavirus. Virol. J. 2014, 11, 82. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Sun, S.; Tai, W.; Chen, J.; Geng, Q.; He, L.; Chen, Y.; Wu, J.; Shi, Z.; et al. Molecular Mechanism for Antibody-Dependent Enhancement of Coronavirus Entry. J. Virol. 2020, 94, e02015-19. [Google Scholar] [CrossRef]
- Liu, L.; Wei, Q.; Lin, Q.; Fang, J.; Wang, H.; Kwok, H.; Tang, H.; Nishiura, K.; Peng, J.; Tan, Z.; et al. Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight 2019, 4, e123158. [Google Scholar] [CrossRef]
- Ricke, D.O. Two Different Antibody-Dependent Enhancement (ADE) Risks for SARS-CoV-2 Antibodies. Front. Immunol. 2021, 12, 640093. [Google Scholar] [CrossRef]
- Maemura, T.; Kuroda, M.; Armbrust, T.; Yamayoshi, S.; Halfmann, P.J.; Kawaoka, Y. Antibody-Dependent Enhancement of SARS-CoV-2 Infection Is Mediated by the IgG Receptors FcγRIIA and FcγRIIIA but Does Not Contribute to Aberrant Cytokine Production by Macrophages. mBio 2021, 12, e0198721. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Edwards, R.J.; Manne, K.; Martinez, D.R.; Schäfer, A.; Alam, S.M.; Wiehe, K.; Lu, X.; Parks, R.; Sutherland, L.L.; et al. In vitro and in vivo functions of SARS-CoV-2 infection-enhancing and neutralizing antibodies. Cell 2021, 184, 4203–4219.e32. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Yan, R.; Liu, M.; Liu, Z.; Wang, Y.; Luan, D.; Wu, K.; Song, Z.; Sun, T.; Ma, Y. Antibody-dependent enhancement (ADE) of SARS-CoV-2 infection in recovered COVID-19 patients: Studies based on cellular and structural biology analysis. medRxiv 2020. [Google Scholar] [CrossRef]
- Cross, R.W.; Prasad, A.N.; Borisevich, V.; Woolsey, C.; Agans, K.N.; Deer, D.J.; Dobias, N.S.; Geisbert, J.B.; Fenton, K.A.; Geisbert, T.W. Use of convalescent serum reduces severity of COVID-19 in nonhuman primates. Cell Rep. 2021, 34, 108837. [Google Scholar] [CrossRef] [PubMed]
- Joyner, M.J.; Bruno, K.A.; Klassen, S.A.; Kunze, K.L.; Johnson, P.W.; Lesser, E.R.; Wiggins, C.C.; Senefeld, J.W.; Klompas, A.M.; Hodge, D.O.; et al. Safety update: COVID-19 convalescent plasma in 20,000 hospitalized patients. Mayo Clin. Proc. 2020, 95, 1888–1897. [Google Scholar] [CrossRef] [PubMed]
- Arvin, A.M.; Fink, K.; Schmid, M.A.; Cathcart, A.; Spreafico, R.; Havenar-Daughton, C.; Lanzavecchia, A.; Corti, D.; Virgin, H.W. A perspective on potential antibody-dependent enhancement of SARS-CoV-2. Nature 2020, 584, 353–363. [Google Scholar] [CrossRef]
- Liu, Y.; Soh, W.T.; Kishikawa, J.-I.; Hirose, M.; Nakayama, E.E.; Li, S.; Sasai, M.; Suzuki, T.; Tada, A.; Arakawa, A.; et al. An infectivity-enhancing site on the SARS-CoV-2 spike protein targeted by antibodies. Cell 2021, 184, 3452–3466.e18. [Google Scholar] [CrossRef]
- Loo, Y.-M.; McTamney, P.M.; Arends, R.H.; Abram, M.E.; Aksyuk, A.A.; Diallo, S.; Flores, D.J.; Kelly, E.J.; Ren, K.; Roque, R.; et al. The SARS-CoV-2 monoclonal antibody combination, AZD7442, is protective in non-human primates and has an extended half-life in humans. Sci. Transl. Med. 2022, 14, eabl8124. [Google Scholar] [CrossRef]
- Cathcart, A.L.; Havenar-Daughton, C.; Lempp, F.A.; Ma, D.; Schmid, M.A.; Agostini, M.L.; Guarino, B.; Di iulio, J.; Rosen, L.E.; Tucker, H.; et al. The dual function monoclonal antibodies VIR-7831 and VIR-7832 demonstrate potent in vitro and in vivo activity against SARS-CoV-2. bioRxiv 2021. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Fact Sheet for Health Care Providers: Emergency Use Authorization (EUA) of Bamlanivimab. Available online: https://www.fda.gov/media/143603/download (accessed on 1 June 2021).
- Woolsey, C.; Borisevich, V.; Prasad, A.N.; Agans, K.N.; Deer, D.J.; Dobias, N.S.; Heymann, J.C.; Foster, S.L.; Levine, C.B.; Medina, L.; et al. Establishment of an African green monkey model for COVID-19 and protection against re-infection. Nat. Immunol. 2021, 22, 86–98. [Google Scholar] [CrossRef]
- Blair, R.V.; Vaccari, M.; Doyle-Meyers, L.A.; Roy, C.J.; Russell-Lodrigue, K.; Fahlberg, M.; Monjure, C.J.; Beddingfield, B.; Plante, K.S.; Plante, J.A.; et al. Acute Respiratory Distress in Aged, SARS-CoV-2–Infected African Green Monkeys but Not Rhesus Macaques. Am. J. Pathol. 2021, 191, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Hartman, A.L.; Nambulli, S.; McMillen, C.M.; White, A.G.; Tilston-Lunel, N.; Albe, J.R.; Cottle, E.L.; Dunn, M.D.; Frye, L.J.; Gilliland, T.H.; et al. SARS-CoV-2 infection of African green monkeys results in mild respiratory disease discernible by PET/CT imaging and prolonged shedding of infectious virus from both respiratory and gastrointestinal tracts. PLoS Pathog. 2020, 16, e1008903. [Google Scholar] [CrossRef] [PubMed]
- Cross, R.W.; Agans, K.N.; Prasad, A.N.; Borisevich, V.; Woolsey, C.; Deer, D.J.; Dobias, N.S.; Geisbert, J.B.; Fenton, K.A.; Geisbert, T.W. Intranasal exposure of African green monkeys to SARS-CoV-2 results in acute phase pneumonia with shedding and lung injury still present in the early convalescence phase. Virol. J. 2020, 17, 125. [Google Scholar] [CrossRef] [PubMed]
- R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 1 June 2021).
- Rubin, D.B. Multiple Imputation for Nonresponse in Surveys; John Wiley & Sons: New York, NY, USA, 1987; ISBN 9780470316696. [Google Scholar]
- Barnard, J.; Rubin, D. Small-sample degrees of freedom with multiple imputation. Biometrika 1999, 86, 948–955. [Google Scholar] [CrossRef]
- McGonagle, D.; Sharif, K.; O’Regan, A.; Bridgewood, C. The role of cytokines including interleukin-6 in COVID-19 induced pneumonia and macrophage activation syndrome-like disease. Autoimmune Rev. 2020, 19, 102537. [Google Scholar] [CrossRef] [PubMed]
- Thulin, N.K.; Brewer, R.C.; Sherwood, R.; Bournazos, S.; Edwards, K.G.; Ramadoss, N.S.; Taubenberger, J.K.; Memoli, M.; Gentles, A.J.; Jagannathan, P.; et al. Maternal anti-dengue IgG fucosylation predicts susceptibility to dengue disease in infants. Cell Rep. 2020, 31, 107642. [Google Scholar] [CrossRef]
- Taylor, A.; Foo, S.-S.; Bruzzone, R.; Dinh, L.V.; King, N.J.C.; Mahalingam, S. Fc receptors in antibody-dependent enhancement of viral infections. Immunol. Rev. 2015, 268, 340–364. [Google Scholar] [CrossRef]
- Cardozo, T.; Veazey, R. Informed consent disclosure to vaccine trial subjects of risk of COVID-19 vaccines worsening clinical disease. Int. J. Clin. Pract. 2021, 75, e13795. [Google Scholar] [CrossRef]
- Li, D.; Edwards, R.J.; Manne, K.; Martinez, D.R.; Schafer, A.; Alam, S.M.; Wiehe, K.; Lu, X.; Parks, R.; Sutherland, L.L.; et al. The functions of SARS-CoV-2 neutralizing and infection-enhancing antibodies in vitro and in mice and nonhuman primates. bioRxiv 2021. [Google Scholar] [CrossRef]
- Lombardi, A.; Bozzi, G.; Ungaro, R.; Villa, S.; Castelli, V.; Mangioni, D.; Muscatello, A.; Gori, A.; Bandera, A. Mini Review Immunological Consequences of Immunization with COVID-19 mRNA Vaccines: Preliminary Results. Front. Immunol. 2021, 12, 657711. [Google Scholar] [CrossRef]
- Powell, J.; Piszczatoski, C.R. Finally, an FDA Approval for an Immunization Against COVID-19: Hope on the Horizon. Ann. Pharmacother. 2022, 56, 1058–1064. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, A.; Muecksch, F.; Lorenzi, J.C.; Leist, S.R.; Cipolla, M.; Bournazos, S.; Schmidt, F.; Maison, R.M.; Gazumyan, A.; Martinez, D.R.; et al. Antibody potency, effector function, and combinations in protection and therapy for SARS-CoV-2 infection in vivo. J. Exp. Med. 2021, 218, e20201993. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration. Fact Sheet for Health Care Providers: Emergency Use Authorization (EUA) of Caririvimab and Imdevimab. Available online: https://www.regeneron.com/downloads/treatment-covid19-eua-fact-sheet-for-hcp.pdf (accessed on 1 June 2021).
- RECOVERY Collaborative Group. Convalescent plasma in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet 2021, 397, 2049–2059. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Voysey, M.; Clemens, S.A.C.; Madhi, S.A.; Weckx, L.Y.; Folegatti, P.M.; Aley, P.K.; Angus, B.; Baillie, V.L.; Barnabas, S.L.; Bhorat, Q.E.; et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: An interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet 2021, 397, 99–111. [Google Scholar] [CrossRef]
- Sadoff, J.; Le Gars, M.; Shukarev, G.; Heerwegh, D.; Truyers, C.; de Groot, A.M.; Stoop, J.; Tete, S.; Van Damme, W.; Leroux-Roels, I.; et al. Interim Results of a Phase 1–2a Trial of Ad26. COV2. S COVID-19 Vaccine. N. Engl. J. Med. 2021, 384, 1824–1835. [Google Scholar] [CrossRef]
- Weinreich, D.M.; Sivapalasingam, S.; Norton, T.; Ali, S.; Gao, H.; Bhore, R.; Musser, B.J.; Soo, Y.; Rofail, D.; Im, J.; et al. REGN-COV2, a Neutralizing Antibody Cocktail, in Outpatients with COVID-19. N. Engl. J. Med. 2020, 384, 238–251. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Incidence (Total Lobes Affected) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Male | Female | |||||||
| Control | Bamlanivimab | Control | Bamlanivimab | |||||
| Dose (mg/kg) | 0.05 | 20 | 0.05 | 20 | 0.05 | 20 | 0.05 | 20 |
| Inflammation, Mixed Cell, Bronchi/Bronchioles | ||||||||
| Minimal | 3 | 3 | 8 | 2 | 7 | 2 | 3 | 0 |
| Mild | 11 | 10 | 2 | 10 | 2 | 6 | 10 | 7 |
| Moderate | 3 | 5 | 4 | 1 | 1 | 1 | 2 | 0 |
| Marked | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Increased Cells, Alveolar/Septal | ||||||||
| Minimal | 2 | 2 | 1 | 3 | 4 | 1 | 0 | 4 |
| Mild | 12 | 13 | 10 | 15 | 12 | 10 | 17 | 14 |
| Moderate | 4 | 1 | 5 | 0 | 0 | 1 | 0 | 0 |
| Marked | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| Congestion/Hemorrhage, Alveolar | ||||||||
| Minimal | 8 | 7 | 3 | 8 | 0 | 3 | 3 | 4 |
| Mild | 2 | 7 | 8 | 2 | 6 | 2 | 12 | 8 |
| Moderate | 0 | 3 | 5 | 0 | 4 | 0 | 0 | 0 |
| Marked | 0 | 0 | 0 | 0 | 2 | 1 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cross, R.W.; Wiethoff, C.M.; Brown-Augsburger, P.; Berens, S.; Blackbourne, J.; Liu, L.; Wu, X.; Tetreault, J.; Dodd, C.; Sina, R.; et al. The Therapeutic Monoclonal Antibody Bamlanivimab Does Not Enhance SARS-CoV-2 Infection by FcR-Mediated Mechanisms. Pathogens 2023, 12, 1408. https://doi.org/10.3390/pathogens12121408
Cross RW, Wiethoff CM, Brown-Augsburger P, Berens S, Blackbourne J, Liu L, Wu X, Tetreault J, Dodd C, Sina R, et al. The Therapeutic Monoclonal Antibody Bamlanivimab Does Not Enhance SARS-CoV-2 Infection by FcR-Mediated Mechanisms. Pathogens. 2023; 12(12):1408. https://doi.org/10.3390/pathogens12121408
Chicago/Turabian StyleCross, Robert W., Christopher M. Wiethoff, Patricia Brown-Augsburger, Shawn Berens, Jamie Blackbourne, Ling Liu, Xiaohua Wu, Jonathan Tetreault, Carter Dodd, Ramtin Sina, and et al. 2023. "The Therapeutic Monoclonal Antibody Bamlanivimab Does Not Enhance SARS-CoV-2 Infection by FcR-Mediated Mechanisms" Pathogens 12, no. 12: 1408. https://doi.org/10.3390/pathogens12121408