
Epidemiology, Molecular Pathogenesis, Immuno-Pathogenesis, Immune Escape Mechanisms and Vaccine Evaluation for HPV-Associated Carcinogenesis
 ,
,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Epidemiology of HPV-Induced Cancers
3. HPV Molecular Structure and Classification
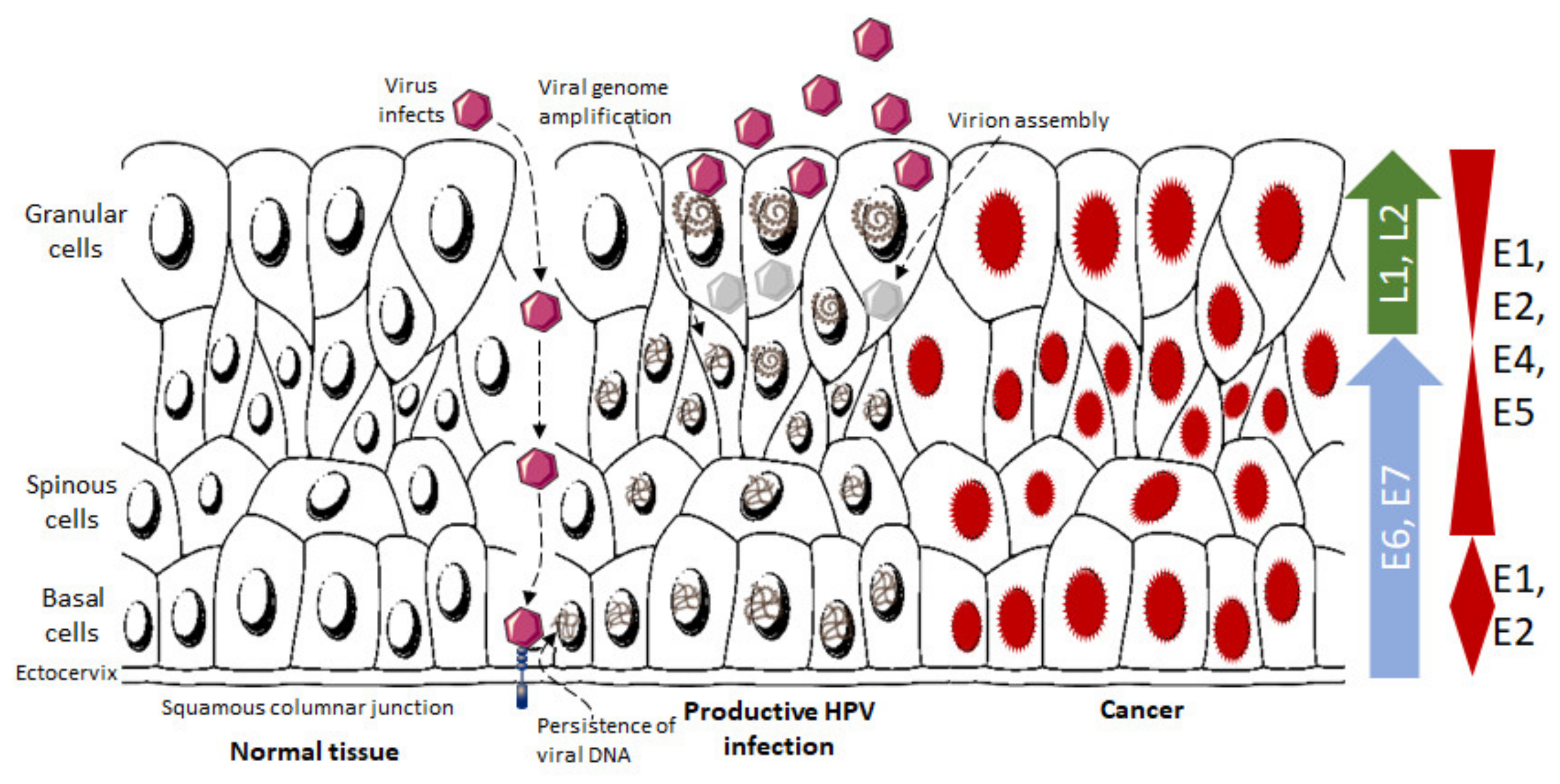
4. HPV Infection and Its Role in Cancer Progression
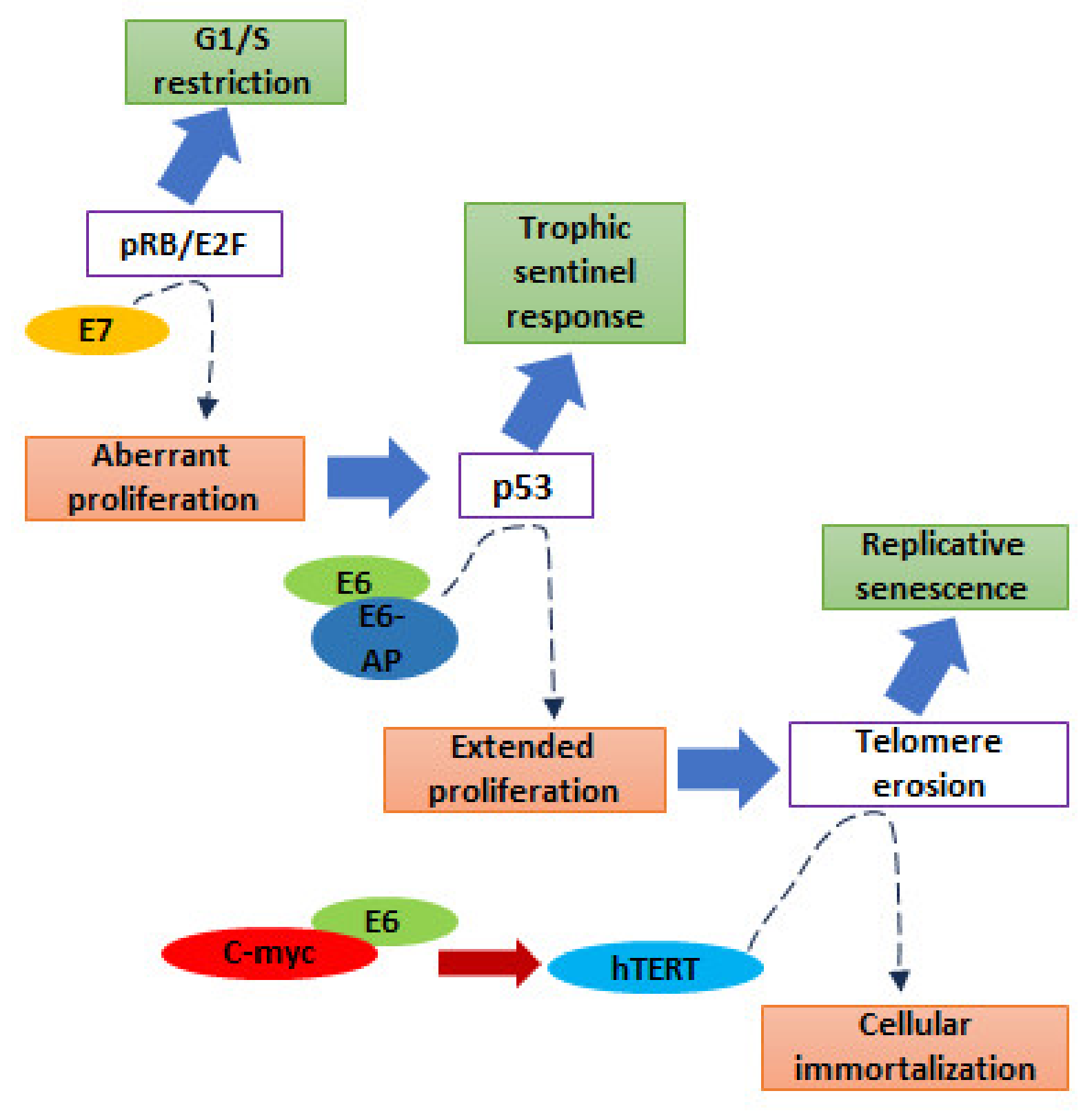
5. Molecular and Genetic Basis of HPV-Induced Carcinogenesis
5.1. E7 Protein
5.2. E6 Protein
6. HPV Infection Generates Host Immune Response
7. Immune Escape Mechanism for HPV Perseverance
8. Prevention Strategies to Control HPV-Associated Cancers
- (A) Screening:
- (B) Vaccination:
- (C) Treatment:
9. Vaccination Stratagems to Fight against HPV Infection
10. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hathaway, J.K. Hpv: Diagnosis, prevention, and treatment. Clin. Obstet. Gynecol. 2012, 55, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Álvarez, M.I.; Gómez-Urquiza, J.L.; Husein-El Ahmed, H.; Albendín-García, L.; Gómez-Salgado, J.; Cañadas-De la Fuente, G.A. Prevalence and risk factors of human papillomavirus in male patients: A systematic review and meta-analysis. Int. J. Environ. Res. Public Health 2018, 15, 2210. [Google Scholar] [CrossRef] [PubMed]
- Kombe Kombe, A.J.; Li, B.; Zahid, A.; Mengist, H.M.; Bounda, G.-A.; Zhou, Y.; Jin, T. Epidemiology and burden of human papillomavirus and related diseases, molecular pathogenesis, and vaccine evaluation. Front. Public Health 2021, 8, 552028. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Herrero, R.; Bosetti, C.; Munoz, N.; Bosch, F.X.; Eluf-Neto, J.; Castellsagué, X.; Meijer, C.J.; Van den Brule, A.J.; Franceschi, S. Herpes simplex virus-2 as a human papillomavirus cofactor in the etiology of invasive cervical cancer. J. Natl. Cancer Inst. 2002, 94, 1604–1613. [Google Scholar] [CrossRef]
- Muñoz, N.; Castellsagué, X.; de González, A.B.; Gissmann, L. Hpv in the etiology of human cancer. Vaccine 2006, 24 (Suppl. S3), S1–S10. [Google Scholar] [CrossRef]
- Romero, J.; Girón, C.; Marina, V. Hormonal contraception as a risk factor for cancer prevention: Biological evidence, immunological and epidemiological. Ginecol. Obstet. Mex. 2011, 79, 533–539. [Google Scholar]
- Jensen, K.; Schmiedel, S.; Norrild, B.; Frederiksen, K.; Iftner, T.; Kjaer, S. Parity as a cofactor for high-grade cervical disease among women with persistent human papillomavirus infection: A 13-year follow-up. Br. J. Cancer 2013, 108, 234–239. [Google Scholar] [CrossRef]
- Luhn, P.; Walker, J.; Schiffman, M.; Zuna, R.E.; Dunn, S.T.; Gold, M.A.; Smith, K.; Mathews, C.; Allen, R.A.; Zhang, R. The role of co-factors in the progression from human papillomavirus infection to cervical cancer. Gynecol. Oncol. 2013, 128, 265–270. [Google Scholar] [CrossRef]
- Deligeoroglou, E.; Giannouli, A.; Athanasopoulos, N.; Karountzos, V.; Vatopoulou, A.; Dimopoulos, K.; Creatsas, G. Hpv infection: Immunological aspects and their utility in future therapy. Infect. Dis. Obstet. Gynecol. 2013, 2013, 540850. [Google Scholar] [CrossRef]
- Sasagawa, T.; Takagi, H.; Makinoda, S. Immune responses against human papillomavirus (hpv) infection and evasion of host defense in cervical cancer. J. Infect. Chemother. 2012, 18, 807–815. [Google Scholar] [CrossRef]
- Mao, C.; Hughes, J.P.; Kiviat, N.; Kuypers, J.; Lee, S.-K.; Adam, D.E.; Koutsky, L.A. Clinical findings among young women with genital human papillomavirus infection. Am. J. Obstet. Gynecol. 2003, 188, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Ault, K.A. Epidemiology and natural history of human papillomavirus infections in the female genital tract. Infect. Dis. Obstet. Gynecol. 2006, 2006, 040470. [Google Scholar] [CrossRef] [PubMed]
- Schröer, N.; Pahne, J.; Walch, B.; Wickenhauser, C.; Smola, S. Molecular pathobiology of human cervical high-grade lesions: Paracrine stat3 activation in tumor-instructed myeloid cells drives local mmp-9 expression. Cancer Res. 2011, 71, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Walch-Rückheim, B.; Mavrova, R.; Henning, M.; Vicinus, B.; Kim, Y.-J.; Bohle, R.M.; Juhasz-Böss, I.; Solomayer, E.-F.; Smola, S. Stromal fibroblasts induce ccl20 through il6/c/ebpβ to support the recruitment of th17 cells during cervical cancer progression. Cancer Res. 2015, 75, 5248–5259. [Google Scholar] [CrossRef]
- Pahne-Zeppenfeld, J.; Schröer, N.; Walch-Rückheim, B.; Oldak, M.; Gorter, A.; Hegde, S.; Smola, S. Cervical cancer cell-derived interleukin-6 impairs ccr7-dependent migration of mmp-9-expressing dendritic cells. Int. J. Cancer 2014, 134, 2061–2073. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Simard, E.P.; Dorell, C.; Noone, A.-M.; Markowitz, L.E.; Kohler, B.; Eheman, C.; Saraiya, M.; Bandi, P.; Saslow, D. Annual report to the nation on the status of cancer, 1975–2009, featuring the burden and trends in human papillomavirus (hpv)–associated cancers and hpv vaccination coverage levels. J. Natl. Cancer Inst. 2013, 105, 175–201. [Google Scholar] [CrossRef] [PubMed]
- Bruni, L.; Barrionuevo-Rosas, L.; Albero, G.; Serrano, B.; Mena, M.; Gómez, D.; Muñoz, J.; Bosch, F.; De Sanjosé, S. Human Papillomavirus and Related Diseases Report; ICO/IARC Information Centre on HPV and Cancer (HPV Information Centre): Barcelona, Spain, 2019; p. 307. [Google Scholar]
- Bruni, L.; Albero, G.; Serrano, B.; Mena, M.; Collado, J.; Gómez, D.; Muñoz, J.; Bosch, F.; de Sanjosé, S. Human Papillomavirus and Related Diseases in the World. Summary Report 22 October 2021; ICO/IARC Information Centre on HPV and Cancer (HPV Information Centre): Barcelona, Spain, 2023. [Google Scholar]
- Forman, D.; de Martel, C.; Lacey, C.J.; Soerjomataram, I.; Lortet-Tieulent, J.; Bruni, L.; Vignat, J.; Ferlay, J.; Bray, F.; Plummer, M. Global burden of human papillomavirus and related diseases. Vaccine 2012, 30, F12–F23. [Google Scholar] [CrossRef]
- Bruni, L.; Diaz, M.; Castellsagué, M.; Ferrer, E.; Bosch, F.X.; de Sanjosé, S. Cervical human papillomavirus prevalence in 5 continents: Meta-analysis of 1 million women with normal cytological findings. J. Infect. Dis. 2010, 202, 1789–1799. [Google Scholar] [CrossRef]
- Alizon, S.; Murall, C.L.; Bravo, I.G. Why human papillomavirus acute infections matter. Viruses 2017, 9, 293. [Google Scholar] [CrossRef]
- Tan, S.C.; Ismail, M.P.; Duski, D.R.; Othman, N.H.; Ankathil, R. Prevalence and type distribution of human papillomavirus (hpv) in malaysian women with and without cervical cancer: An updated estimate. Biosci. Rep. 2018, 38, BSR20171268. [Google Scholar] [CrossRef]
- Cogliano, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F. Carcinogenicity of human papillomaviruses. Lancet Oncol. 2005, 6, 204. [Google Scholar] [CrossRef]
- De Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef]
- De Martel, C.; Plummer, M.; Vignat, J.; Franceschi, S. Worldwide burden of cancer attributable to hpv by site, country and hpv type. Int. J. Cancer 2017, 141, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, S.; Herrero, R.; Clifford, G.M.; Snijders, P.J.; Arslan, A.; Anh, P.T.H.; Bosch, F.X.; Ferreccio, C.; Hieu, N.T.; Lazcano-Ponce, E. Variations in the age-specific curves of human papillomavirus prevalence in women worldwide. Int. J. Cancer 2006, 119, 2677–2684. [Google Scholar] [CrossRef]
- Arbyn, M.; Weiderpass, E.; Bruni, L.; de Sanjosé, S.; Saraiya, M.; Ferlay, J.; Bray, F. Estimates of incidence and mortality of cervical cancer in 2018: A worldwide analysis. Lancet Glob. Health 2020, 8, e191–e203. [Google Scholar] [CrossRef] [PubMed]
- Trottier, H.; Franco, E.L. The epidemiology of genital human papillomavirus infection. Vaccine 2006, 24 (Suppl. S1), S4–S15. [Google Scholar] [CrossRef]
- Johnson, C.; Obanor, N.; DeWeese, A. Human papillomavirus and cancer in men. Health Sci. J. 2016, 10, 1. [Google Scholar] [CrossRef]
- Patel, P.; Bush, T.; Kojic, E.M.; Conley, L.; Unger, E.R.; Darragh, T.M.; Henry, K.; Hammer, J.; Escota, G.; Palefsky, J.M. Prevalence, incidence, and clearance of anal high-risk human papillomavirus infection among hiv-infected men in the sun study. J. Infect. Dis. 2018, 217, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, H.; Li, X.; Yang, Y.; Xin, H.; Li, M.; Feng, B.; Gao, L. Anal human papillomavirus genotyping among hiv-positive men who have sex with men in Xi’an, China. PLoS ONE 2015, 10, e0125120. [Google Scholar] [CrossRef]
- Moreira Jr, E.D.; Giuliano, A.R.; Palefsky, J.; Flores, C.A.; Goldstone, S.; Ferris, D.; Hillman, R.J.; Moi, H.; Stoler, M.H.; Marshall, B. Incidence, clearance, and disease progression of genital human papillomavirus infection in heterosexual men. J. Infect. Dis. 2014, 210, 192–199. [Google Scholar] [CrossRef]
- Giuliano, A.R.; Lazcano-Ponce, E.; Villa, L.L.; Flores, R.; Salmeron, J.; Lee, J.-H.; Papenfuss, M.R.; Abrahamsen, M.; Jolles, E.; Nielson, C.M. The human papillomavirus infection in men study: Human papillomavirus prevalence and type distribution among men residing in Brazil, Mexico, and the United States. Cancer Epidemiol. Biomark. Prev. 2008, 17, 2036–2043. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Senapati, R.; Nayak, B.; Kar, S.K.; Dwibedi, B. HPV genotypes distribution in Indian women with and without cervical carcinoma: Implication for HPV vaccination program in Odisha, eastern India. BMC Infect. Dis. 2017, 17, 30. [Google Scholar] [CrossRef]
- Mehrotra, R.; Yadav, K. Cervical cancer: Formulation and implementation of govt of India guidelines for screening and management. Indian J. Gynecol. Oncol. 2022, 20, 4. [Google Scholar] [CrossRef] [PubMed]
- Pilleron, S.; Cabasag, C.J.; Ferlay, J.; Bray, F.; Luciani, S.; Almonte, M.; Piñeros, M. Cervical cancer burden in Latin America and the Caribbean: Where are we? Int. J. Cancer 2020, 147, 1638–1648. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Strategy towards Eliminating Cervical Cancer as a Public Health Problem; WHO: Geneva, Switzerland, 2019. [Google Scholar]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- De Sanjose, S.; Brotons, M.; Pavon, M.A. The natural history of human papillomavirus infection. Baillieres Best Pract. Res. Clin. Obstet. Gynaecol. 2018, 47, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Wójcik, L.; Samulak, D.; Makowska, M.; Romanowicz, H.; Kojs, Z.; Smolarz, B.; Michalska, M. The role of human papillomavirus in cervical cancer. Int. J. Cancer Clin. Res. 2019, 6, 125. [Google Scholar]
- Graham, S.V. The human papillomavirus replication cycle, and its links to cancer progression: A comprehensive review. Clin. Sci. 2017, 131, 2201–2221. [Google Scholar] [CrossRef]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30, F55–F70. [Google Scholar] [CrossRef]
- Wang, X.; Huang, X.; Zhang, Y. Involvement of human papillomaviruses in cervical cancer. Front. Microbiol. 2018, 9, 2896. [Google Scholar] [CrossRef] [PubMed]
- Bernard, H.-U.; Burk, R.D.; Chen, Z.; Van Doorslaer, K.; Zur Hausen, H.; de Villiers, E.-M. Classification of papillomaviruses (pvs) based on 189 pv types and proposal of taxonomic amendments. Virology 2010, 401, 70–79. [Google Scholar] [CrossRef] [PubMed]
- De Villiers, E.-M. Cross-roads in the classification of papillomaviruses. Virology 2013, 445, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Gottschling, M.; Göker, M.; Köhler, A.; Lehmann, M.D.; Stockfleth, E.; Nindl, I. Cutaneotropic human β-/γ-papillomaviruses are rarely shared between family members. J. Investig. Dermatol. 2009, 129, 2427–2434. [Google Scholar] [CrossRef] [PubMed]
- Bottalico, D.; Chen, Z.; Dunne, A.; Ostoloza, J.; McKinney, S.; Sun, C.; Schlecht, N.F.; Fatahzadeh, M.; Herrero, R.; Schiffman, M. The Oral Cavity Contains Abundant Known and Novel Human Papillomaviruses from the Betapapillomavirus and Gammapapillomavirus Genera. J. Infect. Dis. 2011, 204, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, N.; Bosch, F.X.; De Sanjosé, S.; Herrero, R.; Castellsagué, X.; Shah, K.V.; Snijders, P.J.; Meijer, C.J. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 2003, 348, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Tulay, P.; Serakinci, N. The role of human papillomaviruses in cancer progression. J. Cancer Metastasis Treat. 2016, 2, 201. [Google Scholar] [CrossRef]
- Middleton, K.; Peh, W.; Southern, S.; Griffin, H.; Sotlar, K.; Nakahara, T.; El-Sherif, A.; Morris, L.; Seth, R.; Hibma, M. Organization of human papillomavirus productive cycle during neoplastic progression provides a basis for selection of diagnostic markers. J. Virol. 2003, 77, 10186–10201. [Google Scholar] [CrossRef]
- Stanley, M. Pathology and epidemiology of HPV infection in females. Gynecol. Oncol. 2010, 117, S5–S10. [Google Scholar] [CrossRef]
- Beutner, K.R.; Tyring, S. Human papillomavirus and human disease. Am. J. Med. 1997, 102, 9–15. [Google Scholar] [CrossRef]
- Cason, J. Perinatal acquisition of cervical cancer-associated papillomaviruses. BJOG Int. J. Obstet. Gynaecol. 1996, 103, 853–858. [Google Scholar] [CrossRef]
- Favre, M.; Majewski, S.; De Jesus, N.; Malejczyk, M.; Orth, G.; Jablonska, S. A possible vertical transmission of human papillomavirus genotypes associated with epidermodysplasia verruciformis. J. Investig. Dermatol. 1998, 111, 333–336. [Google Scholar] [CrossRef]
- Bosch, F.X.; Qiao, Y.L.; Castellsagué, X. CHAPTER 2 the epidemiology of human papillomavirus infection and its association with cervical cancer. Int. J. Gynecol. Obstet. 2006, 94, S8–S21. [Google Scholar] [CrossRef] [PubMed]
- Newell, K.A. Chapter 23—Wound closure. In Essential Clinical Procedures, 2nd ed.; Dehn, R.W., Asprey, D.P., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2007; pp. 313–341. [Google Scholar]
- Doorbar, J. The papillomavirus life cycle. J. Clin. Virol. 2005, 32, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Narisawa-Saito, M.; Kiyono, T. Basic mechanisms of high-risk human papillomavirus-induced carcinogenesis: Roles of e6 and e7 proteins. Cancer Sci. 2007, 98, 1505–1511. [Google Scholar] [CrossRef] [PubMed]
- Herfs, M.; Yamamoto, Y.; Laury, A.; Wang, X.; Nucci, M.R.; McLaughlin-Drubin, M.E.; Münger, K.; Feldman, S.; McKeon, F.D.; Xian, W. A discrete population of squamocolumnar junction cells implicated in the pathogenesis of cervical cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 10516–10521. [Google Scholar] [CrossRef] [PubMed]
- Egawa, K. Do human papillomaviruses target epidermal stem cells? Dermatology 2003, 207, 251–254. [Google Scholar] [CrossRef]
- Pyeon, D.; Pearce, S.M.; Lank, S.M.; Ahlquist, P.; Lambert, P.F. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog. 2009, 5, e1000318. [Google Scholar] [CrossRef]
- Raff, A.B.; Woodham, A.W.; Raff, L.M.; Skeate, J.G.; Yan, L.; Da Silva, D.M.; Schelhaas, M.; Kast, W.M. The evolving field of human papillomavirus receptor research: A review of binding and entry. J. Virol. 2013, 87, 6062–6072. [Google Scholar] [CrossRef]
- Abban, C.Y.; Meneses, P.I. Usage of heparan sulfate, integrins, and FAK in HPV16 infection. Virology 2010, 403, 1–16. [Google Scholar] [CrossRef]
- Surviladze, Z.; Dziduszko, A.; Ozbun, M.A. Essential roles for soluble virion-associated heparan sulfonated proteoglycans and growth factors in human papillomavirus infections. PLoS Pathog. 2012, 8, e1002519. [Google Scholar] [CrossRef]
- Culp, T.D.; Budgeon, L.R.; Marinkovich, M.P.; Meneguzzi, G.; Christensen, N.D. Keratinocyte-secreted laminin 5 can function as a transient receptor for human papillomaviruses by binding virions and transferring them to adjacent cells. J. Virol. 2006, 80, 8940–8950. [Google Scholar] [CrossRef]
- Dziduszko, A.; Ozbun, M.A. Annexin a2 and s100a10 regulate human papillomavirus type 16 entry and intracellular trafficking in human keratinocytes. J. Virol. 2013, 87, 7502–7515. [Google Scholar] [CrossRef]
- Schäfer, G.; Graham, L.M.; Lang, D.M.; Blumenthal, M.J.; Bergant Marušič, M.; Katz, A.A. Vimentin modulates infectious internalization of human papillomavirus 16 pseudovirions. J. Virol. 2017, 91, e00307-17. [Google Scholar] [CrossRef]
- Scheffer, K.D.; Gawlitza, A.; Spoden, G.A.; Zhang, X.A.; Lambert, C.; Berditchevski, F.; Florin, L. Tetraspanin cd151 mediates papillomavirus type 16 endocytosis. J. Virol. 2013, 87, 3435–3446. [Google Scholar] [CrossRef]
- Shafti-Keramat, S.; Handisurya, A.; Kriehuber, E.; Meneguzzi, G.; Slupetzky, K.; Kirnbauer, R. Different heparan sulfate proteoglycans serve ascellular receptors for humanpapillomaviruses. J. Virol. 2003, 77, 13125–13135. [Google Scholar] [CrossRef]
- Schiller, J.T.; Day, P.M.; Kines, R.C. Current understanding of the mechanism of hpv infection. Gynecol. Oncol. 2010, 118, S12–S17. [Google Scholar] [CrossRef]
- Bienkowska-Haba, M.; Patel, H.D.; Sapp, M. Target cell cyclophilins facilitate human papillomavirus type 16 infection. PLoS Pathog. 2009, 5, e1000524. [Google Scholar] [CrossRef]
- Richards, R.M.; Lowy, D.R.; Schiller, J.T.; Day, P.M. Cleavage of the papillomavirus minor capsid protein, l2, at a furin consensus site is necessary for infection. Proc. Natl. Acad. Sci. USA 2006, 103, 1522–1527. [Google Scholar] [CrossRef]
- Kines, R.C.; Thompson, C.D.; Lowy, D.R.; Schiller, J.T.; Day, P.M. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc. Natl. Acad. Sci. USA 2009, 106, 20458–20463. [Google Scholar] [CrossRef]
- DiGiuseppe, S.; Bienkowska-Haba, M.; Guion, L.G.; Sapp, M. Cruising the cellular highways: How human papillomavirus travels from the surface to the nucleus. Virus Res. 2017, 231, 1–9. [Google Scholar] [CrossRef]
- Day, P.M.; Thompson, C.D.; Schowalter, R.M.; Lowy, D.R.; Schiller, J.T. Identification of a role for the trans-golgi network in human papillomavirus 16 pseudovirus infection. J. Virol. 2013, 87, 3862–3870. [Google Scholar] [CrossRef]
- Zhang, W.; Kazakov, T.; Popa, A.; DiMaio, D. Vesicular trafficking of incoming human papillomavirus 16 to the golgi apparatus and endoplasmic reticulum requires γ-secretase activity. mBio 2014, 5, 10–1128. [Google Scholar] [CrossRef]
- DiGiuseppe, S.; Luszczek, W.; Keiffer, T.R.; Bienkowska-Haba, M.; Guion, L.G.; Sapp, M.J. Incoming human papillomavirus type 16 genome resides in a vesicular compartment throughout mitosis. Proc. Natl. Acad. Sci. USA 2016, 113, 6289–6294. [Google Scholar] [CrossRef]
- Day, P.M.; Baker, C.C.; Lowy, D.R.; Schiller, J.T. Establishment of papillomavirus infection is enhanced by promyelocytic leukemia protein (pml) expression. Proc. Natl. Acad. Sci. USA 2004, 101, 14252–14257. [Google Scholar] [CrossRef]
- Everett, R.D. The spatial organization of DNA virus genomes in the nucleus. PLoS Pathog. 2013, 9, e1003386. [Google Scholar] [CrossRef]
- Doorbar, J. Molecular biology of human papillomavirus infection and cervical cancer. Clin. Sci. 2006, 110, 525–541. [Google Scholar] [CrossRef]
- Ozbun, M.A. Human papillomavirus type 31b infection of human keratinocytes and the onset of early transcription. J. Virol. 2002, 76, 11291–11300. [Google Scholar] [CrossRef]
- McBride, A.A. The papillomavirus e2 proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef]
- Sanders, C.M.; Stenlund, A. Transcription factor-dependent loading of the e1 initiator reveals modular assembly of the papillomavirus origin melting complex. J. Biol. Chem. 2000, 275, 3522–3534. [Google Scholar] [CrossRef]
- Chiang, C.-M.; Ustav, M.; Stenlund, A.; Ho, T.F.; Broker, T.R.; Chow, L.T. Viral e1 and e2 proteins support replication of homologous and heterologous papillomaviral origins. Proc. Natl. Acad. Sci. USA 1992, 89, 5799–5803. [Google Scholar] [CrossRef]
- Dreer, M.; van de Poel, S.; Stubenrauch, F. Control of viral replication and transcription by the papillomavirus e8^ e2 protein. Virus Res. 2017, 231, 96–102. [Google Scholar] [CrossRef]
- You, J.; Croyle, J.L.; Nishimura, A.; Ozato, K.; Howley, P.M. Interaction of the bovine papillomavirus e2 protein with brd4 tethers the viral DNA to host mitotic chromosomes. Cell 2004, 117, 349–360. [Google Scholar] [CrossRef]
- Donaldson, M.M.; Boner, W.; Morgan, I.M. Topbp1 regulates human papillomavirus type 16 e2 interaction with chromatin. J. Virol. 2007, 81, 4338–4342. [Google Scholar] [CrossRef]
- Parish, J.L.; Bean, A.M.; Park, R.B.; Androphy, E.J. Chlr1 is required for loading papillomavirus e2 onto mitotic chromosomes and viral genome maintenance. Mol. Cell 2006, 24, 867–876. [Google Scholar] [CrossRef]
- Yu, T.; Peng, Y.-C.; Androphy, E.J. Mitotic kinesin-like protein 2 binds and colocalizes with papillomavirus e2 during mitosis. J. Virol. 2007, 81, 1736–1745. [Google Scholar] [CrossRef]
- Smith, J.A.; Haberstroh, F.S.; White, E.A.; Livingston, D.M.; DeCaprio, J.A.; Howley, P.M. Smcx and components of the tip60 complex contribute to e2 regulation of the hpv e6/e7 promoter. Virology 2014, 468, 311–321. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Lee, A.-Y.; Hou, S.Y.; Kemper, J.K.; Erdjument-Bromage, H.; Tempst, P.; Chiang, C.-M. Brd4 links chromatin targeting to hpv transcriptional silencing. Genes Dev. 2006, 20, 2383–2396. [Google Scholar] [CrossRef]
- Westrich, J.A.; Warren, C.J.; Pyeon, D. Evasion of host immune defenses by human papillomavirus. Virus Res. 2017, 231, 21–33. [Google Scholar] [CrossRef]
- Oldak, M.; Smola, H.; Aumailley, M.; Rivero, F.; Pfister, H.; Smola-Hess, S. The human papillomavirus type 8 e2 protein suppresses β4-integrin expression in primary human keratinocytes. J. Virol. 2004, 78, 10738–10746. [Google Scholar] [CrossRef]
- Graham, S.V. Human papillomavirus: Gene expression, regulation and prospects for novel diagnostic methods and antiviral therapies. Future Microbiol. 2010, 5, 1493–1506. [Google Scholar] [CrossRef]
- Thomas, J.T.; Hubert, W.G.; Ruesch, M.N.; Laimins, L.A. Human papillomavirus type 31 oncoproteins e6 and e7 are required for the maintenance of episomes during the viral life cycle in normal human keratinocytes. Proc. Natl. Acad. Sci. USA 1999, 96, 8449–8454. [Google Scholar] [CrossRef]
- Park, R.B.; Androphy, E.J. Genetic analysis of high-risk e6 in episomal maintenance of human papillomavirus genomes in primary human keratinocytes. J. Virol. 2002, 76, 11359–11364. [Google Scholar] [CrossRef]
- Roman, A.; Munger, K. The papillomavirus e7 proteins. Virology 2013, 445, 138–168. [Google Scholar] [CrossRef]
- Huh, K.; Zhou, X.; Hayakawa, H.; Cho, J.-Y.; Libermann, T.A.; Jin, J.; Wade Harper, J.; Munger, K. Human papillomavirus type 16 e7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. J. Virol. 2007, 81, 9737–9747. [Google Scholar] [CrossRef]
- Helt, A.-M.; Galloway, D.A. Destabilization of the retinoblastoma tumor suppressor by human papillomavirus type 16 e7 is not sufficient to overcome cell cycle arrest in human keratinocytes. J. Virol. 2001, 75, 6737–6747. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, W.; Roman, A. The e7 proteins of low-and high-risk human papillomaviruses share the ability to target the prb family member p130 for degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 437–442. [Google Scholar] [CrossRef]
- Hwang, S.G.; Lee, D.; Kim, J.; Seo, T.; Choe, J. Human papillomavirus type 16 e7 binds to e2f1 and activates e2f1-driven transcription in a retinoblastoma protein-independent manner. J. Biol. Chem. 2002, 277, 2923–2930. [Google Scholar] [CrossRef]
- Stoler, M.H.; Wolinsky, S.M.; Whitbeck, A.; Broker, T.R.; Chow, L.T. Differentiation-linked human papillomavirus types 6 and 11 transcription in genital condylomata revealed by in situ hybridization with message-specific RNA probes. Virology 1989, 172, 331–340. [Google Scholar] [CrossRef]
- Beyer-Finkler, E.; Stoler, M.H.; Girardi, F.; Pfister, H. Cell differentiation-related gene expression of human papillomavirus 33. Med. Microbiol. Immunol. 1990, 179, 185–192. [Google Scholar] [CrossRef]
- Van Doorslaer, K.; Li, Z.; Xirasagar, S.; Maes, P.; Kaminsky, D.; Liou, D.; Sun, Q.; Kaur, R.; Huyen, Y.; McBride, A.A. The papillomavirus episteme: A major update to the papillomavirus sequence database. Nucleic Acids Res. 2017, 45, D499–D506. [Google Scholar] [CrossRef]
- Baker, C.C.; Phelps, W.C.; Lindgren, V.; Braun, M.J.; Gonda, M.A.; Howley, P.M. Structural and transcriptional analysis of human papillomavirus type 16 sequences in cervical carcinoma cell lines. J. Virol. 1987, 61, 962–971. [Google Scholar] [CrossRef]
- Münger, K.; Baldwin, A.; Edwards, K.M.; Hayakawa, H.; Nguyen, C.L.; Owens, M.; Grace, M.; Huh, K. Mechanisms of human papillomavirus-induced oncogenesis. J. Virol. 2004, 78, 11451–11460. [Google Scholar] [CrossRef]
- Rizzolio, F.; Lucchetti, C.; Caligiuri, I.; Marchesi, I.; Caputo, M.; Klein-Szanto, A.; Bagella, L.; Castronovo, M.; Giordano, A. Retinoblastoma tumor-suppressor protein phosphorylation and inactivation depend on direct interaction with pin1. Cell Death Differ. 2012, 19, 1152–1161. [Google Scholar] [CrossRef]
- Howley, P.M. Warts, cancer and ubiquitylation: Lessons from the papillomaviruses. Trans. Am. Clin. Climatol. Assoc. 2006, 117, 113. [Google Scholar]
- Münger, K.; Basile, J.R.; Duensing, S.; Eichten, A.; Gonzalez, S.L.; Grace, M.; Zacny, V.L. Biological activities and molecular targets of the human papillomavirus e7 oncoprotein. Oncogene 2001, 20, 7888–7898. [Google Scholar] [CrossRef]
- Sen, P.; Ganguly, P.; Ganguly, N. Modulation of DNA methylation by human papillomavirus e6 and e7 oncoproteins in cervical cancer. Oncol. Lett. 2018, 15, 11–22. [Google Scholar] [CrossRef]
- Yin, F.-F.; Wang, N.; Bi, X.-N.; Yu, X.; Xu, X.-H.; Wang, Y.-L.; Zhao, C.-Q.; Luo, B.; Wang, Y.-K. Serine/threonine kinases 31 (stk31) may be a novel cellular target gene for the hpv16 oncogene e7 with potential as a DNA hypomethylation biomarker in cervical cancer. Virol. J. 2016, 13, 60. [Google Scholar] [CrossRef]
- Dueñas-González, A.; Lizano, M.; Candelaria, M.; Cetina, L.; Arce, C.; Cervera, E. Epigenetics of cervical cancer. An overview and therapeutic perspectives. Mol. Cancer 2005, 4, 38. [Google Scholar] [CrossRef]
- Patel, D.; Huang, S.-M.; Baglia, L.A.; McCance, D.J. The e6 protein of human papillomavirus type 16 binds to and inhibits co-activation by cbp and p300. EMBO J. 1999, 18, 5061–5072. [Google Scholar] [CrossRef]
- Zimmermann, H.; Degenkolbe, R.; Bernard, H.-U.; O’Connor, M.J. The human papillomavirus type 16 e6 oncoprotein can down-regulate p53 activity by targeting the transcriptional coactivator cbp/p300. J. Virol. 1999, 73, 6209–6219. [Google Scholar] [CrossRef]
- Thomas, M.; Massimi, P.; Jenkins, J.; Banks, L. Hpv-18 e6 mediated inhibition of p53 DNA binding activity is independent of e6 induced degradation. Oncogene 1995, 10, 261–268. [Google Scholar]
- Georgescu, S.R.; Mitran, C.I.; Mitran, M.I.; Caruntu, C.; Sarbu, M.I.; Matei, C.; Nicolae, I.; Tocut, S.M.; Popa, M.I.; Tampa, M. New insights in the pathogenesis of hpv infection and the associated carcinogenic processes: The role of chronic inflammation and oxidative stress. J. Immunol. Res. 2018, 2018, 5315816. [Google Scholar] [CrossRef]
- Giannini, S.L.; Hubert, P.; Doyen, J.; Boniver, J.; Delvenne, P. Influence of the mucosal epithelium microenvironment on langerhans cells: Implications for the development of squamous intraepithelial lesions of the cervix. Int. J. Cancer 2002, 97, 654–659. [Google Scholar] [CrossRef]
- Leong, C.M.; Doorbar, J.; Nindl, I.; Yoon, H.S.; Hibma, M.H. Deregulation of e-cadherin by human papillomavirus is not confined to high-risk, cancer-causing types. Br. J. Dermatol. 2010, 163, 1253–1263. [Google Scholar] [CrossRef]
- Fausch, S.C.; Da Silva, D.M.; Kast, W.M. Differential uptake and cross-presentation of human papillomavirus virus-like particles by dendritic cells and langerhans cells. Cancer Res. 2003, 63, 3478–3482. [Google Scholar]
- Lebre, M.C.; van der Aar, A.M.; van Baarsen, L.; van Capel, T.M.; Schuitemaker, J.H.; Kapsenberg, M.L.; de Jong, E.C. Human keratinocytes express functional toll-like receptor 3, 4, 5, and 9. J. Investig. Dermatol. 2007, 127, 331–341. [Google Scholar] [CrossRef]
- Karim, R.; Meyers, C.; Backendorf, C.; Ludigs, K.; Offringa, R.; van Ommen, G.-J.B.; Melief, C.J.; van der Burg, S.H.; Boer, J.M. Human papillomavirus deregulates the response of a cellular network comprising of chemotactic and proinflammatory genes. PLoS ONE 2011, 6, e17848. [Google Scholar] [CrossRef]
- Stanley, M. Immune responses to human papillomavirus and the development of human papillomavirus vaccines. In Human Papillomavirus; Elsevier: Amsterdam, The Netherlands, 2020; pp. 283–297. [Google Scholar]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef]
- DeCarlo, C.A.; Rosa, B.; Jackson, R.; Niccoli, S.; Escott, N.G.; Zehbe, I. Toll-like receptor transcriptome in the hpv-positive cervical cancer microenvironment. J. Immunol. Res. 2012, 2012, 785825. [Google Scholar] [CrossRef] [PubMed]
- Hasan, U.A.; Bates, E.; Takeshita, F.; Biliato, A.; Accardi, R.; Bouvard, V.; Mansour, M.; Vincent, I.; Gissmann, L.; Iftner, T. Tlr9 expression and function is abolished by the cervical cancer-associated human papillomavirus type 16. J. Immunol. 2007, 178, 3186–3197. [Google Scholar] [CrossRef] [PubMed]
- DeCarlo, C.A.; Severini, A.; Edler, L.; Escott, N.G.; Lambert, P.F.; Ulanova, M.; Zehbe, I. Ifn-κ, a novel type i ifn, is undetectable in hpv-positive human cervical keratinocytes. Lab. Investig. 2010, 90, 1482–1491. [Google Scholar] [CrossRef] [PubMed]
- Rincon-Orozco, B.; Halec, G.; Rosenberger, S.; Muschik, D.; Nindl, I.; Bachmann, A.; Ritter, T.M.; Dondog, B.; Ly, R.; Bosch, F.X. Epigenetic silencing of interferon-κ in human papillomavirus type 16–positive cells. Cancer Res. 2009, 69, 8718–8725. [Google Scholar] [CrossRef] [PubMed]
- Routes, J.M.; Morris, K.; Ellison, M.C.; Ryan, S. Macrophages kill human papillomavirus type 16 e6-expressing tumor cells by tumor necrosis factor alpha-and nitric oxide-dependent mechanisms. J. Virol. 2005, 79, 116–123. [Google Scholar] [CrossRef]
- Hacke, K.; Rincon-Orozco, B.; Buchwalter, G.; Siehler, S.Y.; Wasylyk, B.; Wiesmüller, L.; Rösl, F. Regulation of mcp-1 chemokine transcription by p53. Mol. Cancer 2010, 9, 82. [Google Scholar] [CrossRef]
- Guess, J.C.; McCance, D.J. Decreased migration of langerhans precursor-like cells in response to human keratinocytes expressing human papillomavirus type 16 e6/e7 is related to reduced macrophage inflammatory protein-3α production. J. Virol. 2005, 79, 14852–14862. [Google Scholar] [CrossRef]
- Garcia-Iglesias, T.; del Toro-Arreola, A.; Albarran-Somoza, B.; del Toro-Arreola, S.; Sanchez-Hernandez, P.E.; Ramirez-Dueñas, M.G.; Balderas-Peña, L.M.A.; Bravo-Cuellar, A.; Ortiz-Lazareno, P.C.; Daneri-Navarro, A. Low nkp30, nkp46 and nkg2d expression and reduced cytotoxic activity on nk cells in cervical cancer and precursor lesions. BMC Cancer 2009, 9, 186. [Google Scholar] [CrossRef]
- Laffort, C.; Le Deist, F.; Favre, M.; Caillat-Zucman, S.; Radford-Weiss, I.; Fraitag, S.; Blanche, S.; Cavazzana-Calvo, M.; de Saint Basile, G.; de Villartay, J.P. Severe cutaneous papillomavirus disease after haemopoietic stem-cell transplantation in patients with severe combined immune deficiency caused by common γc cytokine receptor subunit or jak-3 deficiency. Lancet 2004, 363, 2051–2054. [Google Scholar] [CrossRef]
- Miura, S.; Kawana, K.; Schust, D.J.; Fujii, T.; Yokoyama, T.; Iwasawa, Y.; Nagamatsu, T.; Adachi, K.; Tomio, A.; Tomio, K. Cd1d, a sentinel molecule bridging innate and adaptive immunity, is downregulated by the human papillomavirus (hpv) e5 protein: A possible mechanism for immune evasion by hpv. J. Virol. 2010, 84, 11614–11623. [Google Scholar] [CrossRef]
- Barros, M.R.; de Melo, C.M.L.; Barros, M.L.C.M.G.R.; de Cássia Pereira de Lima, R.; de Freitas, A.C.; Venuti, A. Activities of stromal and immune cells in hpv-related cancers. J. Exp. Clin. Cancer Res. 2018, 37, 137. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Chacon, R.; Velasco-Ramirez, S.F.; Flores-Romo, L.; Daneri-Navarro, A. Immunobiology of hpv infection. Arch. Med. Res. 2009, 40, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J. Host control of human papillomavirus infection and disease. Baillieres Best Pract. Res. Clin. Obstet. Gynaecol. 2018, 47, 27–41. [Google Scholar] [CrossRef] [PubMed]
- De Vos van Steenwijk, P.; Ramwadhdoebe, T.; Goedemans, R.; Doorduijn, E.; Van Ham, J.; Gorter, A.; Van Hall, T.; Kuijjer, M.; Van Poelgeest, M.; Van Der Burg, S. Tumor-infiltrating cd14-positive myeloid cells and cd8-positive t-cells prolong survival in patients with cervical carcinoma. Int. J. Cancer 2013, 133, 2884–2894. [Google Scholar] [CrossRef] [PubMed]
- Masterson, L.; Lechner, M.; Loewenbein, S.; Mohammed, H.; Davies-Husband, C.; Fenton, T.; Sudhoff, H.; Jani, P.; Goon, P.; Sterling, J. Cd8+ t cell response to human papillomavirus 16 e7 is able to predict survival outcome in oropharyngeal cancer. Eur. J. Cancer 2016, 67, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.A.; O’Rorke, M.A.; Wilson, R.; Jamison, J.; Gavin, A.T.; Group, N.I.H.W. Hpv prevalence and type-distribution in cervical cancer and premalignant lesions of the cervix: A population-based study from Northern Ireland. J. Med. Virol. 2016, 88, 1262–1270. [Google Scholar] [CrossRef]
- Steinbach, A.; Riemer, A.B. Immune evasion mechanisms of human papillomavirus: An update. Int. J. Cancer 2018, 142, 224–229. [Google Scholar] [CrossRef]
- Stanley, M.A. Epithelial cell responses to infection with human papillomavirus. Clin. Microbiol. Rev. 2012, 25, 215–222. [Google Scholar] [CrossRef]
- Burgers, W.A.; Blanchon, L.; Pradhan, S.; De Launoit, Y.; Kouzarides, T.; Fuks, F. Viral oncoproteins target the DNA methyltransferases. Oncogene 2007, 26, 1650–1655. [Google Scholar] [CrossRef]
- Nasu, K.; Narahara, H. Pattern recognition via the toll-like receptor system in the human female genital tract. Mediat. Inflamm. 2010, 2010, 976024. [Google Scholar] [CrossRef]
- Evans, M.; Borysiewicz, L.K.; Evans, A.S.; Rowe, M.; Jones, M.; Gileadi, U.; Cerundolo, V.; Man, S. Antigen processing defects in cervical carcinomas limit the presentation of a ctl epitope from human papillomavirus 16 e6. J. Immunol. 2001, 167, 5420–5428. [Google Scholar] [CrossRef]
- Campo, M.; Graham, S.; Cortese, M.; Ashrafi, G.; Araibi, E.; Dornan, E.; Miners, K.; Nunes, C.; Man, S. Hpv-16 e5 down-regulates expression of surface hla class i and reduces recognition by cd8 t cells. Virology 2010, 407, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Hasim, A.; Abudula, M.; Aimiduo, R.; Ma, J.-Q.; Jiao, Z.; Akula, G.; Wang, T.; Abudula, A. Post-transcriptional and epigenetic regulation of antigen processing machinery (APM) components and HLA-I in cervical cancers from Uighur women. PLoS ONE 2012, 7, e44952. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-S.; Kim, E.-J.; Kwon, H.-J.; Hwang, E.-S.; Namkoong, S.-E.; Um, S.-J. Inactivation of interferon regulatory factor-1 tumor suppressor protein by hpv e7 oncoprotein: Implication for the e7-mediated immune evasion mechanism in cervical carcinogenesis. J. Biol. Chem. 2000, 275, 6764–6769. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.; Gray, E.E.; Brunette, R.L.; Stetson, D.B. DNA tumor virus oncogenes antagonize the cgas-sting DNA-sensing pathway. Science 2015, 350, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Hasan, U.A.; Zannetti, C.; Parroche, P.; Goutagny, N.; Malfroy, M.; Roblot, G.; Carreira, C.; Hussain, I.; Müller, M.; Taylor-Papadimitriou, J. The human papillomavirus type 16 e7 oncoprotein induces a transcriptional repressor complex on the toll-like receptor 9 promoter. J. Exp. Med. 2013, 210, 1369–1387. [Google Scholar] [CrossRef]
- Pacini, L.; Savini, C.; Ghittoni, R.; Saidj, D.; Lamartine, J.; Hasan, U.A.; Accardi, R.; Tommasino, M. Downregulation of toll-like receptor 9 expression by beta human papillomavirus 38 and implications for cell cycle control. J. Virol. 2015, 89, 11396–11405. [Google Scholar] [CrossRef] [PubMed]
- Karim, R.; Tummers, B.; Meyers, C.; Biryukov, J.L.; Alam, S.; Backendorf, C.; Jha, V.; Offringa, R.; van Ommen, G.-J.B.; Melief, C.J. Human papillomavirus (hpv) upregulates the cellular deubiquitinase uchl1 to suppress the keratinocyte’s innate immune response. PLoS Pathog. 2013, 9, e1003384. [Google Scholar] [CrossRef]
- Richards, K.H.; Wasson, C.W.; Watherston, O.; Doble, R.; Eric Blair, G.; Wittmann, M.; Macdonald, A. The human papillomavirus (hpv) e7 protein antagonises an imiquimod-induced inflammatory pathway in primary human keratinocytes. Sci. Rep. 2015, 5, 12922. [Google Scholar] [CrossRef]
- Ma, W.; Tummers, B.; Van Esch, E.M.; Goedemans, R.; Melief, C.J.; Meyers, C.; Boer, J.M.; van der Burg, S.H. Human papillomavirus downregulates the expression of ifitm1 and ripk3 to escape from ifnγ-and tnfα-mediated antiproliferative effects and necroptosis. Front. Immunol. 2016, 7, 496. [Google Scholar] [CrossRef]
- Brotherton, J.M.L.; Wheeler, C.; Clifford, G.M.; Elfström, M.; Saville, M.; Kaldor, J.; Machalek, D.A. Surveillance systems for monitoring cervical cancer elimination efforts: Focus on hpv infection, cervical dysplasia, cervical screening and treatment. Prev. Med. 2021, 144, 106293. [Google Scholar] [CrossRef]
- Nishimura, H.; Yeh, P.T.; Oguntade, H.; Kennedy, C.E.; Narasimhan, M. Hpv self-sampling for cervical cancer screening: A systematic review of values and preferences. BMJ Glob. Health 2021, 6, e003743. [Google Scholar] [CrossRef] [PubMed]
- Lucksom, P.G.; Sherpa, M.L.; Pradhan, A.; Lal, S.; Gupta, C. Advances in hpv screening tests for cervical cancer-a review. J. Obstet. Gynaecol. India 2022, 72, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.; Mangwendeza, P.; Maparo, T.; Mhizha, T.; Demke, O.; Murwira, S.; Dickson, H.; Madzima, B.; Khan, S. Comparative analysis between self-collected and clinician-collected samples for hpv testing in public health facilities in Zimbabwe. J. Clin. Virol. 2021, 145, 105017. [Google Scholar] [CrossRef]
- Safaeian, M.; Solomon, D.; Castle, P.E. Cervical cancer prevention--cervical screening: Science in evolution. Obstet. Gynecol. Clin. N. Am. 2007, 34, 739–760. [Google Scholar] [CrossRef] [PubMed]
- Poli, U.R.; Bidinger, P.D.; Gowrishankar, S. Visual inspection with acetic acid (via) screening program: 7 years experience in early detection of cervical cancer and pre-cancers in rural south India. Indian J. Community Med. 2015, 40, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Bruni, L.; Serrano, B.; Roura, E.; Alemany, L.; Cowan, M.; Herrero, R.; Poljak, M.; Murillo, R.; Broutet, N.; Riley, L.M. Cervical cancer screening programmes and age-specific coverage estimates for 202 countries and territories worldwide: A review and synthetic analysis. Lancet Glob. Health 2022, 10, e1115–e1127. [Google Scholar] [CrossRef]
- World Health Organization. WHO Guideline for Screening and Treatment of Cervical Pre-Cancer Lesions for Cervical Cancer Prevention: Use of mRNA Tests for Human Papillomavirus (HPV); World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Dahlstrom, K.R.; Day, A.T.; Sturgis, E.M. Prevention and screening of hpv malignancies. Semin. Radiat. Oncol. 2021, 31, 297–308. [Google Scholar] [CrossRef]
- Illah, O.; Olaitan, A. Updates on HPV vaccination. Diagnostics 2023, 13, 243. [Google Scholar] [CrossRef]
- Dorji, T.; Nopsopon, T.; Tamang, S.T.; Pongpirul, K. Human papillomavirus vaccination uptake in low-and middle-income countries: A meta-analysis. EClinicalMedicine 2021, 34, 100836. [Google Scholar] [CrossRef] [PubMed]
- Kamolratanakul, S.; Pitisuttithum, P. Human papillomavirus vaccine efficacy and effectiveness against cancer. Vaccines 2021, 9, 1413. [Google Scholar] [CrossRef] [PubMed]
- Akhatova, A.; Azizan, A.; Atageldiyeva, K.; Ashimkhanova, A.; Marat, A.; Iztleuov, Y.; Suleimenova, A.; Shamkeeva, S.; Aimagambetova, G. Prophylactic human papillomavirus vaccination: From the origin to the current state. Vaccines 2022, 10, 1912. [Google Scholar] [CrossRef] [PubMed]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L. A review of human carcinogens—Part b: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.J.; Castle, P.E.; Lorincz, A.T.; Wacholder, S.; Sherman, M.; Scott, D.R.; Rush, B.B.; Glass, A.G.; Schiffman, M. The elevated 10-year risk of cervical precancer and cancer in women with human papillomavirus (hpv) type 16 or 18 and the possible utility of type-specific hpv testing in clinical practice. J. Natl. Cancer Inst. 2005, 97, 1072–1079. [Google Scholar] [CrossRef] [PubMed]
- Brisson, M.; Bénard, É.; Drolet, M.; Bogaards, J.A.; Baussano, I.; Vänskä, S.; Jit, M.; Boily, M.-C.; Smith, M.A.; Berkhof, J. Population-level impact, herd immunity, and elimination after human papillomavirus vaccination: A systematic review and meta-analysis of predictions from transmission-dynamic models. Lancet Public Health 2016, 1, e8–e17. [Google Scholar] [CrossRef] [PubMed]
- Papillomavirus, W.O.-D.H. Vaccine offers solid protection against cervical cancer. Saudi Med. J. 2022, 43, 538. [Google Scholar]
- World Health Organization. Human papillomavirus vaccines: WHO position paper, May 2017—Recommendations. Vaccine 2017, 35, 5753–5755. [Google Scholar] [CrossRef]
- Garland, S.M. The australian experience with the human papillomavirus vaccine. Clin. Ther. 2014, 36, 17–23. [Google Scholar] [CrossRef]
- Markowitz, L.E.; Dunne, E.F.; Saraiya, M.; Chesson, H.W.; Curtis, C.R.; Gee, J.; Bocchini, J.A., Jr.; Unger, E.R. Human papillomavirus vaccination: Recommendations of the advisory committee on immunization practices (ACIP). Morb. Mortal. Wkly. Rep. Recomm. Rep. 2014, 63, 1–30. [Google Scholar]
- Ferris, D.; Samakoses, R.; Block, S.L.; Lazcano-Ponce, E.; Restrepo, J.A.; Reisinger, K.S.; Mehlsen, J.; Chatterjee, A.; Iversen, O.-E.; Sings, H.L. Long-term study of a quadrivalent human papillomavirus vaccine. Pediatrics 2014, 134, e657–e665. [Google Scholar] [CrossRef]
- Goldstone, S.E.; Giuliano, A.R.; Palefsky, J.M.; Lazcano-Ponce, E.; Penny, M.E.; Cabello, R.E.; Moreira, E.D.; Baraldi, E.; Jessen, H.; Ferenczy, A. Efficacy, immunogenicity, and safety of a quadrivalent hpv vaccine in men: Results of an open-label, long-term extension of a randomised, placebo-controlled, phase 3 trial. Lancet Infect. Dis. 2022, 22, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Drolet, M.; Bénard, É.; Boily, M.-C.; Ali, H.; Baandrup, L.; Bauer, H.; Beddows, S.; Brisson, J.; Brotherton, J.M.; Cummings, T. Population-level impact and herd effects following human papillomavirus vaccination programmes: A systematic review and meta-analysis. Lancet Infect. Dis. 2015, 15, 565–580. [Google Scholar] [CrossRef] [PubMed]
- Laurent, J.S.; Luckett, R.; Feldman, S. Hpv vaccination and the effects on rates of hpv-related cancers. Curr. Probl. Cancer 2018, 42, 493–506. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Accelerating the Elimination of Cervical Cancer as a Global Public Health Problem; World Health Organization, Regional Office for South-East Asia: New Delphi, India, 2019. [Google Scholar]
- Vujovich-Dunn, C.; Wand, H.; Brotherton, J.; Gidding, H.; Sisnowski, J.; Lorch, R.; Veitch, M.; Sheppeard, V.; Effler, P.; Skinner, S. Measuring school level attributable risk to support school-based hpv vaccination programs. BMC Public Health 2022, 22, 822. [Google Scholar] [CrossRef] [PubMed]
- Simms, K.T.; Steinberg, J.; Caruana, M.; Smith, M.A.; Lew, J.-B.; Soerjomataram, I.; Castle, P.E.; Bray, F.; Canfell, K. Impact of scaled up human papillomavirus vaccination and cervical screening and the potential for global elimination of cervical cancer in 181 countries, 2020–2099: A modelling study. Lancet Oncol. 2019, 20, 394–407. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.T.; Simms, K.T.; Lew, J.-B.; Smith, M.A.; Brotherton, J.M.; Saville, M.; Frazer, I.H.; Canfell, K. The projected timeframe until cervical cancer elimination in australia: A modelling study. Lancet Public Health 2019, 4, e19–e27. [Google Scholar] [CrossRef] [PubMed]
- Tjalma, W.; Brasseur, C.; Top, G.; Ribesse, N.; Morales, I.; Van Damme, P. Hpv vaccination coverage in the federal state of belgium according to regions and their impact. Facts Views Vis. ObGyn 2018, 10, 101. [Google Scholar] [PubMed]
- Wemrell, M.; Gunnarsson, L. Attitudes toward hpv vaccination in sweden: A survey study. Front. Public Health 2022, 10, 729497. [Google Scholar] [CrossRef]
- Wang, J.; Ploner, A.; Sparén, P.; Lepp, T.; Roth, A.; Arnheim-Dahlström, L.; Sundström, K. Mode of hpv vaccination delivery and equity in vaccine uptake: A nationwide cohort study. Prev. Med. 2019, 120, 26–33. [Google Scholar] [CrossRef]
- Wentzensen, N.; Chirenje, Z.M.; Prendiville, W. Treatment approaches for women with positive cervical screening results in low-and middle-income countries. Prev. Med. 2021, 144, 106439. [Google Scholar] [CrossRef]
- Canfell, K.; Kim, J.J.; Brisson, M.; Keane, A.; Simms, K.T.; Caruana, M.; Burger, E.A.; Martin, D.; Nguyen, D.T.N.; Bénard, É.; et al. Mortality impact of achieving who cervical cancer elimination targets: A comparative modelling analysis in 78 low-income and lower-middle-income countries. Lancet 2020, 395, 591–603. [Google Scholar] [CrossRef] [PubMed]
- Chuang, L.T.; Feldman, S.; Nakisige, C.; Temin, S.; Berek, J.S. Management and care of women with invasive cervical cancer: Asco resource-stratified clinical practice guideline. J. Clin. Oncol. 2016, 34, 3354–3355. [Google Scholar] [CrossRef] [PubMed]
- Kesic, V.; Carcopino, X.; Preti, M.; Vieira-Baptista, P.; Bevilacqua, F.; Bornstein, J.; Chargari, C.; Cruickshank, M.; Erzeneoglu, E.; Gallio, N. The European society of gynaecological oncology (ESGO), the international society for the study of vulvovaginal disease (ISSVD), the European college for the study of vulval disease (ECSVD), and the European federation for colposcopy (EFC) consensus statement on the management of vaginal intraepithelial neoplasia. Int. J. Gynecol. Cancer 2023, 33, 446–461. [Google Scholar] [PubMed]
- Zhang, L.; Wang, Q.; Zhang, H.; Xie, Y.; Sui, L.; Cong, Q. Screening history in vaginal precancer and cancer: A retrospective study of 2131 cases in China. Cancer Manag. Res. 2021, 13, 8855–8863. [Google Scholar] [CrossRef]
- Kim, M.-K.; Lee, I.H.; Lee, K.H. Clinical outcomes and risk of recurrence among patients with vaginal intraepithelial neoplasia: A comprehensive analysis of 576 cases. J. Gynecol. Oncol. 2018, 29, e6. [Google Scholar] [CrossRef]
- Zeligs, K.P.; Byrd, K.; Tarney, C.M.; Howard, R.S.; Sims, B.D.; Hamilton, C.A.; Stany, M.P. A clinicopathologic study of vaginal intraepithelial neoplasia. Obstet. Gynecol. 2013, 122, 1223–1230. [Google Scholar] [CrossRef]
- Rome, R.; England, P. Management of vaginal intraepithelial neoplasia: A series of 132 cases with long-term follow-up. Int. J. Gynecol. Cancer 2000, 10, 382–390. [Google Scholar] [CrossRef]
- Hodeib, M.; Cohen, J.G.; Mehta, S.; Rimel, B.; Walsh, C.S.; Li, A.J.; Karlan, B.Y.; Cass, I. Recurrence and risk of progression to lower genital tract malignancy in women with high grade vain. Gynecol. Oncol. 2016, 141, 507–510. [Google Scholar] [CrossRef]
- Farmer, E.; Cheng, M.A.; Hung, C.F.; Wu, T.C. Vaccination strategies for the control and treatment of hpv infection and hpv-associated cancer. Recent Results Cancer Res. 2021, 217, 157–195. [Google Scholar] [CrossRef]
- Pollard, A.J.; Bijker, E.M. A guide to vaccinology: From basic principles to new developments. Nat. Rev. Immunol. 2021, 21, 83–100. [Google Scholar] [CrossRef]
- Chabeda, A.; Yanez, R.J.R.; Lamprecht, R.; Meyers, A.E.; Rybicki, E.P.; Hitzeroth, I.I. Therapeutic vaccines for high-risk hpv-associated diseases. Papillomavirus Res. 2018, 5, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Buttaro, C.; Fruehauf, J.H. Engineered E. coli as vehicles for targeted therapeutics. Curr. Gene Ther. 2010, 10, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Le Gouellec, A.; Chauchet, X.; Polack, B.; Buffat, L.; Toussaint, B. Bacterial vectors for active immunotherapy reach clinical and industrial stages. Hum. Vaccin. Immunother. 2012, 8, 1454–1458. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Farmer, E.; Wu, T.; Hung, C.-F. Perspectives for therapeutic hpv vaccine development. J. Biomed. Sci. 2016, 23, 75. [Google Scholar] [CrossRef] [PubMed]
- Guirnalda, P.; Wood, L.; Paterson, Y. Listeria monocytogenes and its products as agents for cancer immunotherapy. Adv. Immunol. 2012, 113, 81–118. [Google Scholar] [PubMed]
- Kim, H.J.; Kim, H.-J. Current status and future prospects for human papillomavirus vaccines. Arch. Pharm. Res 2017, 40, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Cory, L.; Chu, C. Adxs-hpv: A therapeutic listeria vaccination targeting cervical cancers expressing the hpv e7 antigen. Hum. Vaccin. Immunother. 2014, 10, 3190–3195. [Google Scholar] [CrossRef]
- Wallecha, A.; Wood, L.; Pan, Z.-K.; Maciag, P.C.; Shahabi, V.; Paterson, Y. Listeria monocytogenes-derived listeriolysin o has pathogen-associated molecular pattern-like properties independent of its hemolytic ability. Clin. Vaccine Immunol. 2013, 20, 77–84. [Google Scholar] [CrossRef]
- Gunn, G.R.; Zubair, A.; Peters, C.; Pan, Z.-K.; Wu, T.-C.; Paterson, Y. Two listeria monocytogenes vaccine vectors that express different molecular forms of human papilloma virus-16 (hpv-16) e7 induce qualitatively different t cell immunity that correlates with their ability to induce regression of established tumors immortalized by hpv-16. J. Immunol. 2001, 167, 6471–6479. [Google Scholar]
- Lin, C.W.; Lee, J.Y.; Tsao, Y.P.; Shen, C.P.; Lai, H.C.; Chen, S.L. Oral vaccination with recombinant listeria monocytogenes expressing human papillomavirus type 16 e7 can cause tumor growth in mice to regress. Int. J. Cancer 2002, 102, 629–637. [Google Scholar] [CrossRef]
- Verch, T.; Pan, Z.-K.; Paterson, Y. Listeria monocytogenes-based antibiotic resistance gene-free antigen delivery system applicable to other bacterial vectors and DNA vaccines. Infect. Immun. 2004, 72, 6418–6425. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.F.; Paterson, Y. Cd4+ cd25+ regulatory t cells that secrete tgfβ and il-10 are preferentially induced by a vaccine vector. J. Immunother. 2004, 27, 339–346. [Google Scholar] [CrossRef]
- Sewell, D.A.; Shahabi, V.; Gunn III, G.R.; Pan, Z.-K.; Dominiecki, M.E.; Paterson, Y. Recombinant listeria vaccines containing pest sequences are potent immune adjuvants for the tumor-associated antigen human papillomavirus-16 e7. Cancer Res. 2004, 64, 8821–8825. [Google Scholar] [CrossRef] [PubMed]
- Souders, N.C.; Sewell, D.A.; Pan, Z.-K.; Hussain, S.F.; Rodriguez, A.; Wallecha, A.; Paterson, Y. Listeria-based vaccines can overcome tolerance by expanding low avidity cd8+ t cells capable of eradicating a solid tumor in a transgenic mouse model of cancer. Cancer Immun. 2007, 7, 2. [Google Scholar] [PubMed]
- Miles, B.A.; Monk, B.J.; Safran, H.P. Mechanistic insights into adxs11-001 human papillomavirus-associated cancer immunotherapy. Gynecol. Oncol. Res. Pract. 2017, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.-F.; Ma, B.; Monie, A.; Tsen, S.-W.; Wu, T. Therapeutic human papillomavirus vaccines: Current clinical trials and future directions. Expert Opin. Biol. Ther 2008, 8, 421–439. [Google Scholar] [CrossRef] [PubMed]
- Borysiewicz, L.; Fiander, A.; Nimako, M.; Man, S.; Wilkinson, G.W.G.; Westmoreland, D.; Evans, A.; Adams, M.; Stacey, S.N.; Boursnell, M. A recombinant vaccinia virus encoding human papillomavirus types 16 and 18, e6 and e7 proteins as immunotherapy for cervical cancer. Lancet 1996, 347, 1523–1527. [Google Scholar] [CrossRef] [PubMed]
- Corona Gutierrez, C.M.; Tinoco, A.; Navarro, T.; Contreras, M.L.; Cortes, R.R.; Calzado, P.; Reyes, L.; Posternak, R.; Morosoli, G.; Verde, M.L. Therapeutic vaccination with mva e2 can eliminate precancerous lesions (cin 1, cin 2, and cin 3) associated with infection by oncogenic human papillomavirus. Hum. Gene Ther. 2004, 15, 421–431. [Google Scholar] [CrossRef]
- Garcia-Hernandez, E.; Gonzalez-Sanchez, J.; Andrade-Manzano, A.; Contreras, M.; Padilla, S.; Guzman, C.; Jimenez, R.; Reyes, L.; Morosoli, G.; Verde, M. Regression of papilloma high-grade lesions (cin 2 and cin 3) is stimulated by therapeutic vaccination with mva e2 recombinant vaccine. Cancer Gene Ther. 2006, 13, 592–597. [Google Scholar] [CrossRef]
- Vici, P.; Pizzuti, L.; Mariani, L.; Zampa, G.; Santini, D.; Di Lauro, L.; Gamucci, T.; Natoli, C.; Marchetti, P.; Barba, M. Targeting immune response with therapeutic vaccines in premalignant lesions and cervical cancer: Hope or reality from clinical studies. Expert Rev. Vaccines 2016, 15, 1327–1336. [Google Scholar] [CrossRef]
- Rosales, R.; López-Contreras, M.; Rosales, C.; Magallanes-Molina, J.R.; Gonzalez-Vergara, R.; Arroyo-Cazarez, J.M.; Ricardez-Arenas, A.; Del Follo-Valencia, A.; Padilla-Arriaga, S.; Guerrero, M.V.; et al. Regression of human papillomavirus intraepithelial lesions is induced by mva e2 therapeutic vaccine. Hum. Gene Ther. 2014, 25, 1035–1049. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.A.; Farmer, E.; Huang, C.; Lin, J.; Hung, C.-F.; Wu, T.-C. Therapeutic DNA vaccines for human papillomavirus and associated diseases. Hum. Gene Ther. 2018, 29, 971–996. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Wang, X.; Cui, H.; Cheung, Y.; Hu, M.; Zhu, S.; Xie, Y. Human papillomavirus type 16 e7 peptide38–61 linked with an immunoglobulin g fragment provides protective immunity in mice. Gynecol. Oncol. 2005, 96, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Khong, H.; Overwijk, W.W. Adjuvants for peptide-based cancer vaccines. J. Immunother. Cancer 2016, 4, 56. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Chen, W.C.; Liu, Z.; Huang, L. Bryostatin-i: A dendritic cell stimulator for chemokines induction and a promising adjuvant for a peptide based cancer vaccine. Cytokine 2010, 52, 238–244. [Google Scholar] [CrossRef]
- Daftarian, P.; Mansour, M.; Benoit, A.C.; Pohajdak, B.; Hoskin, D.W.; Brown, R.G.; Kast, W.M. Eradication of established hpv 16-expressing tumors by a single administration of a vaccine composed of a liposome-encapsulated ctl-t helper fusion peptide in a water-in-oil emulsion. Vaccine 2006, 24, 5235–5244. [Google Scholar] [CrossRef]
- Wu, C.-Y.; Monie, A.; Pang, X.; Hung, C.-F.; Wu, T. Improving therapeutic hpv peptide-based vaccine potency by enhancing cd4+ t help and dendritic cell activation. J. Biomed. Sci. 2010, 17, 88. [Google Scholar] [CrossRef]
- Zhang, Y.-Q.; Tsai, Y.-C.; Monie, A.; Hung, C.-F.; Wu, T.-C. Carrageenan as an adjuvant to enhance peptide-based vaccine potency. Vaccine 2010, 28, 5212–5219. [Google Scholar] [CrossRef]
- Zwaveling, S.; Mota, S.C.F.; Nouta, J.; Johnson, M.; Lipford, G.B.; Offringa, R.; van der Burg, S.H.; Melief, C.J. Established human papillomavirus type 16-expressing tumors are effectively eradicated following vaccination with long peptides. J. Immunol. 2002, 169, 350–358. [Google Scholar] [CrossRef]
- Lin, K.; Doolan, K.; Hung, C.-F.; Wu, T. Perspectives for preventive and therapeutic hpv vaccines. J. Formos. Med. Assoc. 2010, 109, 4–24. [Google Scholar] [CrossRef]
- Su, J.-H.; Wu, A.; Scotney, E.; Ma, B.; Monie, A.; Hung, C.-F.; Wu, T.-C. Immunotherapy for cervical cancer: Research status and clinical potential. BioDrugs 2010, 24, 109–129. [Google Scholar] [CrossRef] [PubMed]
- de Vos van Steenwijk, P.J.; van Poelgeest, M.I.; Ramwadhdoebe, T.H.; Löwik, M.J.; Berends-van der Meer, D.M.; van der Minne, C.E.; Loof, N.M.; Stynenbosch, L.F.; Fathers, L.M.; Valentijn, A.R.P. The long-term immune response after hpv16 peptide vaccination in women with low-grade pre-malignant disorders of the uterine cervix: A placebo-controlled phase ii study. Cancer Immunol. Immunother. 2014, 63, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Domingos-Pereira, S.; Galliverti, G.; Hanahan, D.; Nardelli-Haefliger, D. Carboplatin/paclitaxel, e7-vaccination and intravaginal cpg as tri-therapy towards efficient regression of genital hpv16 tumors. J. Immunother. Cancer 2019, 7, 122. [Google Scholar] [CrossRef] [PubMed]
- Coleman, H.N.; Greenfield, W.W.; Stratton, S.L.; Vaughn, R.; Kieber, A.; Moerman-Herzog, A.M.; Spencer, H.J.; Hitt, W.C.; Quick, C.M.; Hutchins, L.F. Human papillomavirus type 16 viral load is decreased following a therapeutic vaccination. Cancer Immunol. Immunother. 2016, 65, 563–573. [Google Scholar] [CrossRef]
- Zandberg, D.P.; Rollins, S.; Goloubeva, O.; Morales, R.E.; Tan, M.; Taylor, R.; Wolf, J.S.; Schumaker, L.M.; Cullen, K.J.; Zimrin, A. A phase i dose escalation trial of mage-a3-and hpv16-specific peptide immunomodulatory vaccines in patients with recurrent/metastatic (rm) squamous cell carcinoma of the head and neck (scchn). Cancer Immunol. Immunother. 2015, 64, 367–379. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jain, M.; Yadav, D.; Jarouliya, U.; Chavda, V.; Yadav, A.K.; Chaurasia, B.; Song, M. Epidemiology, Molecular Pathogenesis, Immuno-Pathogenesis, Immune Escape Mechanisms and Vaccine Evaluation for HPV-Associated Carcinogenesis. Pathogens 2023, 12, 1380. https://doi.org/10.3390/pathogens12121380
Jain M, Yadav D, Jarouliya U, Chavda V, Yadav AK, Chaurasia B, Song M. Epidemiology, Molecular Pathogenesis, Immuno-Pathogenesis, Immune Escape Mechanisms and Vaccine Evaluation for HPV-Associated Carcinogenesis. Pathogens. 2023; 12(12):1380. https://doi.org/10.3390/pathogens12121380
Chicago/Turabian StyleJain, Meenu, Dhananjay Yadav, Urmila Jarouliya, Vishal Chavda, Arun Kumar Yadav, Bipin Chaurasia, and Minseok Song. 2023. "Epidemiology, Molecular Pathogenesis, Immuno-Pathogenesis, Immune Escape Mechanisms and Vaccine Evaluation for HPV-Associated Carcinogenesis" Pathogens 12, no. 12: 1380. https://doi.org/10.3390/pathogens12121380