
Participation of Single-Nucleotide Variants in IFNAR1 and IFNAR2 in the Immune Response against SARS-CoV-2 Infection: A Systematic Review


 , and
, and 
Abstract
:1. Introduction
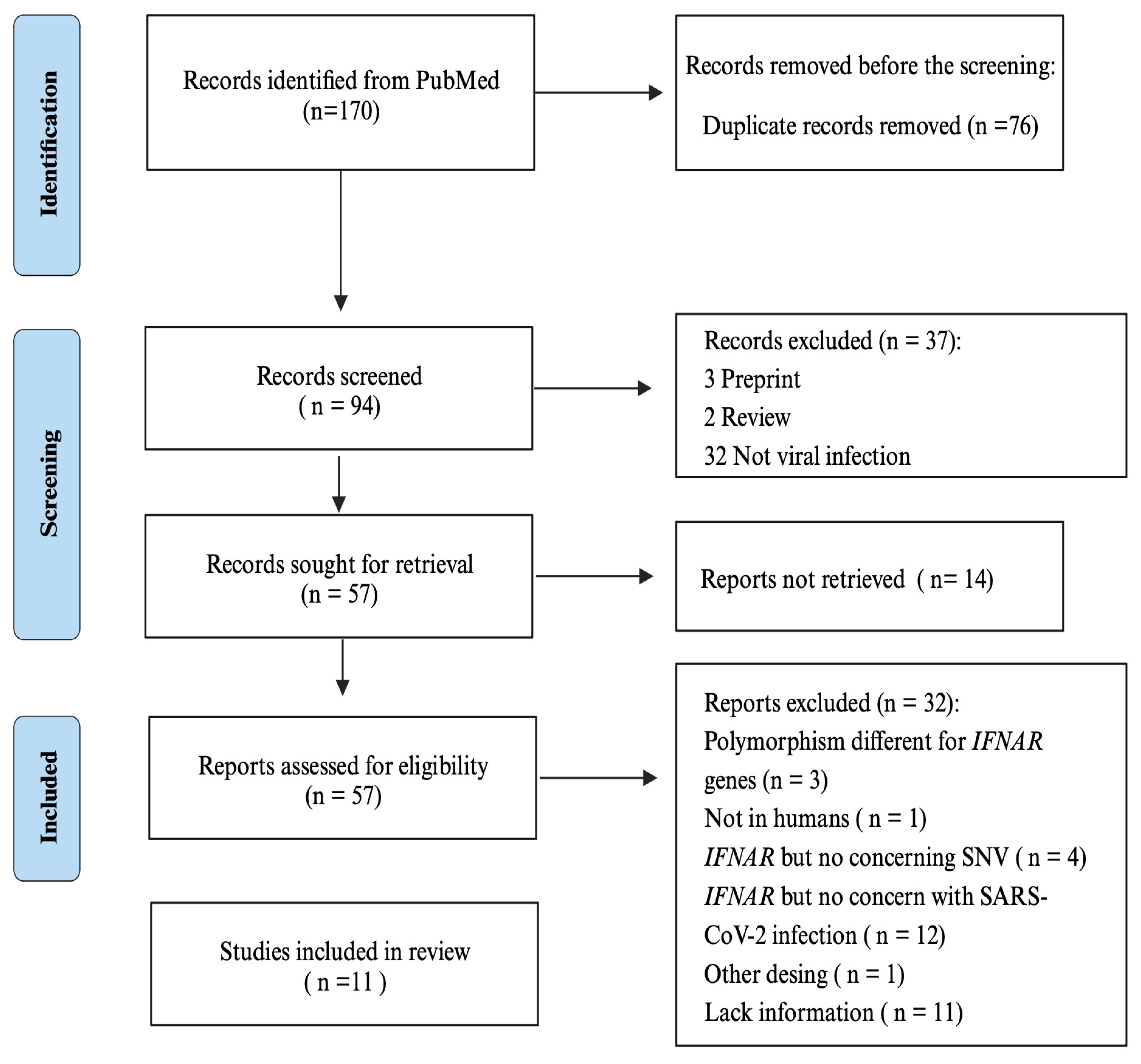
2. Materials and Methods
2.1. Search Strategies
2.2. Inclusion Criteria
2.3. Data Collection
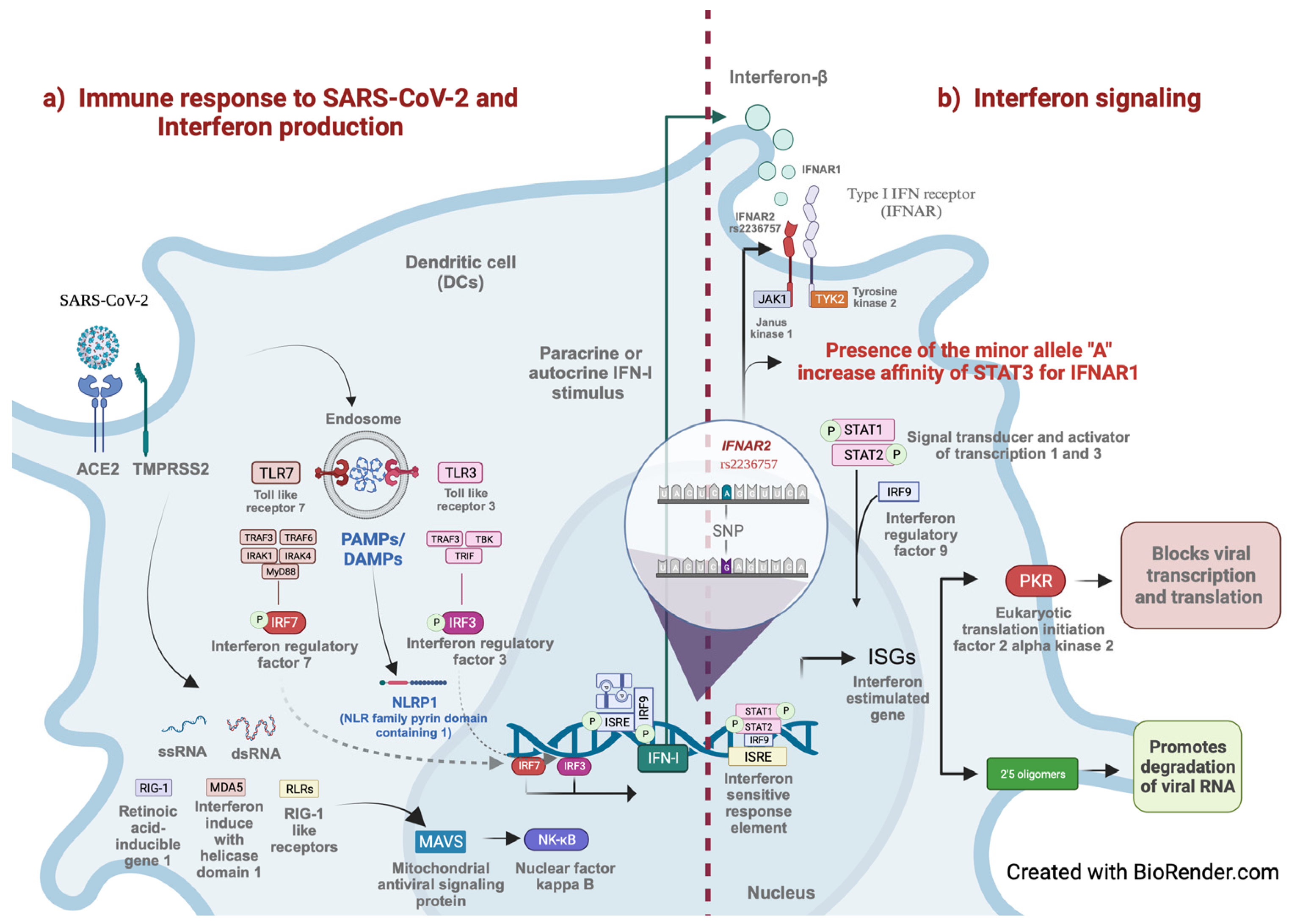
3. Results

{kind=link}
{kind=link}
| Gene | SNV | RA/RG | MAF * (%) | Loci | Gene Consequence | OR | p-Value | Population | Associated Outcome | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| IFNAR2 | rs2236757 | A | 0.34gcc 0.28ukb | 21q22.1 | Intront variant | 1.28 | 4.9 × 10−8 | United Kingdom | [26] | |
| 0.29 | 1.28 | 7.0 × 10−5 | Europe, Africa, South Asia, and Latin America | Severity Mortality | [27] | |||||
| 0.42 | 3.23 | 0.045 | Brazil, non-white | [28] | ||||||
| TMPRSS2 | rs12329760 | T | 0.27 | 21q22.3 | Missense variant | NR | 0.04 | Europe, Africa, South Asia, and Latin America | Severity | [28] |
| ACE1 | rs1799752 | Ins | 0.41 | 17q23.2 | Intront variant | NR | 0.01 | |||
| IFNAR2 | rs2834161 | C | 0.37 | 21q22.1 | Intront variant | 1.25 | 6.10 × 10−7 | Italy, Spain, Norway, Germany, and Australia | Mortality | [29] |
| rs13050728 | T | 0.40 | 21q22.1 | Intront variant | 1.17 | 6.10 × 10−12 | Caucasian | Severity | [31] | |
| rs9976829 | G | 0.33 | 21q22.1 | Intront variant | 1.16 | 2.57 × 10−6 | Caucasian | Susceptibility | [32] | |
| rs2834158 | TC, CC | 0.46 0.18 | 21q22.1 | Intront variant | 1.38 | 0.027 | Mexican | Mortality | [33] | |
| rs3153 | AG, GG | 0.46 0.19 | 21q22.1 | Intront variant | 1.39 | 0.019 | ||||
| rs17860118 | T | 0.18 | 21q22.1 | 5’ UTR | 1.72 | 0.033 | Vietnamese | Susceptibility | [34] | |
| rs2229207 | C | 0.19 | 21q22.1 | Missense variant | 1.89 | 0.012 |
4. Discussion
4.1. Participation of SNVs in IFNAR1 and IFNAR2 in Other Viral Infections
4.2. Perspectives
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Órpez-Zafra, T.; Pavía, J.; Hurtado-Guerrero, I.; Pinto-Medel, M.J.; Bada, J.L.R.; Urbaneja, P.; Suardíaz, M.; Villar, L.M.; Comabella, M.; Montalban, X.; et al. Decreased soluble IFN-β receptor (sIFNAR2) in multiple sclerosis patients: A potential serum diagnostic biomarker. Mult. Scler. J. 2017, 23, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Bastard, P.; Hsiao, K.-C.; Zhang, Q.; Choin, J.; Best, E.; Chen, J.; Gervais, A.; Bizien, L.; Materna, M.; Harmant, C.; et al. A loss-of-function IFNAR1 allele in Polynesia underlies severe viral diseases in homozygotes. J. Exp. Med. 2022, 219, e20220028. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.; Travers, P.; Walport, M. Janeway’s Immunobiology; Garland Science: New York, NY, USA, 2008. [Google Scholar]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Sodeifian, F.; Nikfarjam, M.; Kian, N.; Mohamed, K.; Rezaei, N. The role of type I interferon in the treatment of COVID-19. J. Med Virol. 2022, 94, 63–81. [Google Scholar] [CrossRef] [PubMed]
- Duncan, C.J.; Randall, R.E.; Hambleton, S. Genetic Lesions of Type I Interferon Signalling in Human Antiviral Immunity. Trends Genet. 2021, 37, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bastard, P.; Bolze, A.; Jouanguy, E.; Zhang, S.-Y.; Cobat, A.; Notarangelo, L.D.; Su, H.C.; Abel, L.; Casanova, J.-L. Life-Threatening COVID-19: Defective Interferons Unleash Excessive Inflammation. Med 2020, 1, 14–20. [Google Scholar] [CrossRef]
- Hernandez, N.; Bucciol, G.; Moens, L.; Le Pen, J.; Shahrooei, M.; Goudouris, E.; Shirkani, A.; Changi-Ashtiani, M.; Rokni-Zadeh, H.; Sayar, E.H.; et al. Inherited IFNAR1 deficiency in otherwise healthy patients with adverse reaction to measles and yellow fever live vaccines. J. Exp. Med. 2019, 216, 2057–2070. [Google Scholar] [CrossRef]
- Frodsham, A.J.; Zhang, L.; Dumpis, U.; Taib, N.A.M.; Best, S.; Durham, A.; Hennig, B.J.W.; Hellier, S.; Knapp, S.; Wright, M.; et al. Class II cytokine receptor gene cluster is a major locus for hepatitis B persistence. Proc. Natl. Acad. Sci. USA 2006, 103, 9148–9153. [Google Scholar] [CrossRef] [PubMed]
- Passarelli, C.; Civino, A.; Rossi, M.N.; Cifaldi, L.; Lanari, V.; Moneta, G.M.; Caiello, I.; Bracaglia, C.; Montinaro, R.; Novelli, A.; et al. IFNAR2 Deficiency Causing Dysregulation of NK Cell Functions and Presenting With Hemophagocytic Lymphohistiocytosis. Front. Genet. 2020, 11, 937. [Google Scholar] [CrossRef]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- A Madden, E.; Diamond, M.S. Host cell-intrinsic innate immune recognition of SARS-CoV-2. Curr. Opin. Virol. 2022, 52, 30–38. [Google Scholar] [CrossRef]
- Lowery, S.A.; Sariol, A.; Perlman, S. Innate immune and inflammatory responses to SARS-CoV-2: Implications for COVID-19. Cell Host Microbe 2021, 29, 1052–1062. [Google Scholar] [CrossRef]
- Ahmed, C.M.; Grams, T.R.; Bloom, D.C.; Johnson, H.M.; Lewin, A.S. Individual and Synergistic Anti-Coronavirus Activities of SOCS1/3 Antagonist and Interferon α1 Peptides. Front. Immunol. 2022, 13, 902956. [Google Scholar] [CrossRef]
- Merad, M.; Blish, C.A.; Sallusto, F.; Iwasaki, A. The Immunology and Immunopathology of COVID-19. Science 2022, 375, 1122–1127. Available online: https://www.science.org (accessed on 5 September 2023). [CrossRef]
- Hung, I.F.-N.; Lung, K.-C.; Tso, E.Y.-K.; Liu, R.; Chung, T.W.-H.; Chu, M.-Y.; Ng, Y.-Y.; Lo, J.; Chan, J.; Tam, A.R.; et al. Triple combination of interferon beta-1b, lopinavir–ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: An open-label, randomised, phase 2 trial. Lancet 2020, 395, 1695–1704. [Google Scholar] [CrossRef] [PubMed]
- Fricke-Galindo, I.; Falfán-Valencia, R. Genetics Insight for COVID-19 Susceptibility and Severity: A Review. Front. Immunol. 2021, 12, 622176. [Google Scholar] [CrossRef] [PubMed]
- Carter-Timofte, M.E.; Jørgensen, S.E.; Freytag, M.R.; Thomsen, M.M.; Andersen, N.-S.B.; Al-Mousawi, A.; Hait, A.S.; Mogensen, T.H. Deciphering the Role of Host Genetics in Susceptibility to Severe COVID-19. Front. Immunol. 2020, 11, 1606. [Google Scholar] [CrossRef] [PubMed]
- Lvovs, D.; Favorova, O.O.; Favorov, A.V. A Polygenic Approach to the Study of Polygenic Diseases. Acta Naturae 2012, 4, 59–71. [Google Scholar] [CrossRef]
- The Severe COVID-19 GWAS Group. Genome Association Study of Severe COVID-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; the PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. BMJ 2009, 339, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Russell, C.K.; Gregory, D.M. Evaluation of qualitative research studies. Évid. Based Nurs. 2003, 6, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Aromataris, E.; Fernandez, R.; Godfrey, C.M.; Holly, C.; Khalil, H.; Tungpunkom, P. Summarizing systematic reviews. Int. J. Evid. Based Heal. 2015, 13, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.; Downe, S. Appraising the quality of qualitative research. Midwifery 2006, 22, 108–119. [Google Scholar] [CrossRef]
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Walker, S.; Parkinson, N.; Fourman, M.H.; Russell, C.D.; et al. Genetic mechanisms of critical illness in COVID-19. Nature 2021, 591, 92–98. [Google Scholar] [CrossRef]
- Horowitz, J.E.; Kosmicki, J.A.; Damask, A.; Sharma, D.; Roberts, G.H.L.; Justice, A.E.; Banerjee, N.; Coignet, M.V.; Yadav, A.; Leader, J.B.; et al. Genome-wide analysis provides genetic evidence that ACE2 influences COVID-19 risk and yields risk scores associated with severe disease. Nat. Genet. 2022, 54, 382–392. [Google Scholar] [CrossRef]
- Dieter, C.; Brondani, L.d.A.; Lemos, N.E.; Schaeffer, A.F.; Zanotto, C.; Ramos, D.T.; Girardi, E.; Pellenz, F.M.; Camargo, J.L.; Moresco, K.S.; et al. Polymorphisms in ACE1, TMPRSS2, IFIH1, IFNAR2, and TYK2 Genes Are Associated with Worse Clinical Outcomes in COVID-19. Genes 2023, 14, 29. [Google Scholar] [CrossRef]
- Degenhardt, F.; Ellinghaus, D.; Juzenas, S.; Lerga-Jaso, J.; Wendorff, M.; Maya-Miles, D.; Uellendahl-Werth, F.; ElAbd, H.; Arora, J.; Lenning, O.B.; et al. Detailed stratified GWAS analysis for severe COVID-19 in four European populations. Hum. Mol. Genet. 2022, 31, 3945–3966. [Google Scholar] [CrossRef]
- Liu, D.; Yang, J.; Feng, B.; Lu, W.; Zhao, C.; Li, L. Mendelian randomization analysis identified genes pleiotropically associated with the risk and prognosis of COVID-19. J. Infect. 2021, 82, 126–132. [Google Scholar] [CrossRef]
- Gaziano, L.; Giambartolomei, C.; Pereira, A.C.; Gaulton, A.; Posner, D.C.; Swanson, S.A.; Ho, Y.-L.; Iyengar, S.K.; Kosik, N.M.; Vujkovic, M.; et al. Actionable druggable genome-wide Mendelian randomization identifies repurposing opportunities for COVID-19. Nat. Med. 2021, 27, 668–676. [Google Scholar] [CrossRef]
- Ma, Y.; Huang, Y.; Zhao, S.; Yao, Y.; Zhang, Y.; Qu, J.; Wu, N.; Su, J. Integrative genomics analysis reveals a 21q22.11 locus contributing risk to COVID-19. Hum. Mol. Genet. 2021, 30, 1247–1258. [Google Scholar] [CrossRef] [PubMed]
- Fricke-Galindo, I.; Martínez-Morales, A.; Chávez-Galán, L.; Ocaña-Guzmán, R.; Buendía-Roldán, I.; Pérez-Rubio, G.; Hernández-Zenteno, R.d.J.; Verónica-Aguilar, A.; Alarcón-Dionet, A.; Aguilar-Duran, H.; et al. IFNAR2 relevance in the clinical outcome of individuals with severe COVID-19. Front. Immunol. 2022, 13, 949413. [Google Scholar] [CrossRef] [PubMed]
- Nhung, V.P.; Ton, N.D.; Ngoc, T.T.B.; Thuong, M.T.H.; Hai, N.T.T.; Oanh, K.T.P.; Hien, L.T.T.; Thach, P.N.; Van Hai, N.; Ha, N.H. Host Genetic Risk Factors Associated with COVID-19 Susceptibility and Severity in Vietnamese. Genes 2022, 13, 1884. [Google Scholar] [CrossRef] [PubMed]
- Welzel, T.M.; Morgan, T.R.; Bonkovsky, H.L.; Naishadham, D.; Pfeiffer, R.M.; Wright, E.C.; Hutchinson, A.A.; Crenshaw, A.T.; Bashirova, A.; Carrington, M.; et al. Variants in interferon-alpha pathway genes and response to pegylated interferon-Alpha2a plus ribavirin for treatment of chronic hepatitis C virus infection in the hepatitis C antiviral long-term treatment against cirrhosis trial. Hepatology 2009, 49, 1847–1858. [Google Scholar] [CrossRef]
- Smieszek, S.P.; Polymeropoulos, V.M.; Xiao, C.; Polymeropoulos, C.M.; Polymeropoulos, M.H. Loss-of-function mutations in IFNAR2 in COVID-19 severe infection susceptibility. J. Glob. Antimicrob. Resist. 2021, 26, 239–240. [Google Scholar] [CrossRef]
- Khanmohammadi, S.; Rezaei, N.; Khazaei, M.; Shirkani, A. A Case of Autosomal Recessive Interferon Alpha/Beta Receptor Alpha Chain (IFNAR1) Deficiency with Severe COVID-19. J. Clin. Immunol. 2022, 42, 19–24. [Google Scholar] [CrossRef]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef]
- Duncan, C.J.; Skouboe, M.K.; Howarth, S.; Hollensen, A.K.; Chen, R.; Børresen, M.L.; Thompson, B.J.; Spegarova, J.S.; Hatton, C.F.; Stæger, F.F.; et al. Life-threatening viral disease in a novel form of autosomal recessive IFNAR2 deficiency in the Arctic. J. Exp. Med. 2022, 219, e20212427. [Google Scholar] [CrossRef]
- Jalkanen, J.; Khan, S.; Elima, K.; Huttunen, T.; Wang, N.; Hollmén, M.; Elo, L.L.; Jalkanen, S. Polymorphism in interferon alpha/beta receptor contributes to glucocorticoid response and outcome of ARDS and COVID-19. Crit. Care 2023, 27, 112. [Google Scholar] [CrossRef]
- Raza, R.Z.; Abbasi, S.W. An Evolutionary Insight Into the Heterogeneous Severity Pattern of the SARS-CoV-2 Infection. Front. Genet. 2022, 13, 859508. [Google Scholar] [CrossRef]
- Pahl, M.C.; Le Coz, C.; Su, C.; Sharma, P.; Thomas, R.M.; Pippin, J.A.; Cabrera, E.C.; Johnson, M.E.; Leonard, M.E.; Lu, S.; et al. Implicating effector genes at COVID-19 GWAS loci using promoter-focused Capture-C in disease-relevant immune cell types. Genome Biol. 2022, 23, 125. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Ke, Y.; Shen, W.; Shi, S.; Wang, Y.; Lin, K.; Guo, X.; Wang, C.; Zhang, Y.; Zhao, Z. Targeted screening of genetic associations with COVID-19 susceptibility and severity. Front. Genet. 2022, 13, 1073880. [Google Scholar] [CrossRef] [PubMed]
- Edahiro, R.; Shirai, Y.; Takeshima, Y.; Sakakibara, S.; Yamaguchi, Y.; Murakami, T.; Morita, T.; Kato, Y.; Liu, Y.-C.; Motooka, D.; et al. Single-cell analyses and host genetics highlight the role of innate immune cells in COVID-19 severity. Nat. Genet. 2023, 55, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Akter, S.; Roy, A.S.; Tonmoy, M.I.Q.; Islam, S. Deleterious single nucleotide polymorphisms (SNPs) of human IFNAR2 gene facilitate COVID-19 severity in patients: A comprehensive in silico approach. J. Biomol. Struct. Dyn. 2022, 40, 11173–11189. [Google Scholar] [CrossRef] [PubMed]
- Duncan, C.J.A.; Mohamad, S.M.B.; Young, D.F.; Skelton, A.J.; Leahy, T.R.; Munday, D.C.; Butler, K.M.; Morfopoulou, S.; Brown, J.R.; Hubank, M.; et al. Human IFNAR2 deficiency: Lessons for antiviral immunity. Sci. Transl. Med. 2015, 7, 307ra154. [Google Scholar] [CrossRef]
- Ma, N.; Zhang, X.; Yang, L.; Zhou, J.; Liu, W.; Gao, X.; Yu, F.; Zheng, W.; Ding, S.; Gao, P.; et al. Role of Functional IFNL4, IFNLR1, IFNA, IFNAR2 Polymorphisms in Hepatitis B virus-related liver disease in Han Chinese population. J. Viral Hepat. 2018, 25, 306–313. [Google Scholar] [CrossRef]
- He, X.-X.; Chang, Y.; Jiang, H.-J.; Tang, F.; Meng, F.-Y.; Xie, Q.-H.; Li, P.-Y.; Song, Y.-H.; Lin, J.-S.; Zou, R.; et al. Persistent Effect of IFNAR-1 Genetic Polymorphism on the Long-Term Pathogenesis of Chronic HBV Infection. Available online: www.liebertonline.com (accessed on 6 September 2023).
- Wada, M.; Marusawa, H.; Yamada, R.; Nasu, A.; Osaki, Y.; Kudo, M.; Nabeshima, M.; Fukuda, Y.; Chiba, T.; Matsuda, F. Association of genetic polymorphisms with interferon-induced haematologic adverse effects in chronic hepatitis C patients. J. Viral Hepat. 2009, 16, 388–396. [Google Scholar] [CrossRef]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired Type I Interferon Activity and Inflammatory Responses in Severe COVID-19 Patients. Science 2020, 369, 718–724. Available online: https://www.science.org (accessed on 7 September 2023). [CrossRef]
- Davidson, S.; Maini, M.K.; Wack, A.; Jafarzadeh, A.; Nemati, M.; Saha, B.; Bansode, Y.D.; Jafarzadeh, S. Disease-promoting effects of type i interferons in viral, bacterial, and coinfections. J. Interf. Cytokine Res. 2015, 35, 252–264. [Google Scholar] [CrossRef]
- Prokunina-Olsson, L.; Alphonse, N.; Dickenson, R.E.; Durbin, J.E.; Glenn, J.S.; Hartmann, R.; Kotenko, S.V.; Lazear, H.M.; O’brien, T.R.; Odendall, C.; et al. COVID-19 and emerging viral infections: The case for interferon lambda. J. Exp. Med. 2020, 217, e20200653. [Google Scholar] [CrossRef]
- Hwang, S.Y.; Hertzog, P.J.; Holland, K.A.; Sumarsono, S.H.; Tymms, M.J.; Hamilton, J.A.; Whitty, G.; Bertoncello, I.; Kola, I. A Null Mutation in the Gene Encoding a Type I Interferon Receptor Component Eliminates Antiproliferative and Antiviral Responses to Interferons a and f8 and Alters Macrophage Responses (Gene Targeting/Viral Infection/Macrophages/Signal Transduction). 1995. Available online: https://www.pnas.org (accessed on 12 September 2023).
- Langer, J.A. Interferon at 50: New Molecules, New Potential, New (and Old) Questions Downloaded from. Available online: www.stke.org/cgi/content/full/2007/405/pe53http://stke.sciencemag.org/ (accessed on 12 September 2023).
- Mantlo, E.; Bukreyeva, N.; Maruyama, J.; Paessler, S.; Huang, C. Antiviral activities of type I interferons to SARS-CoV-2 infection. Antivir. Res. 2020, 179, 104811. [Google Scholar] [CrossRef]
- Lokugamage, K.G.; Hage, A.; de Vries, M.; Valero-Jimenez, A.M.; Schindewolf, C.; Dittmann, M.; Rajsbaum, R.; Menachery, V.D. Type I Interferon Susceptibility Distinguishes SARS-CoV-2 from SARS-CoV. J. Virol. 2020, 94, 10–128. [Google Scholar] [CrossRef]
- Busnadiego, I.; Fernbach, S.; Pohl, M.O.; Karakus, U.; Huber, M.; Trkola, A.; Stertz, S.; Hale, B.G. Antiviral activity of type i, ii, and iii interferons counterbalances ace2 inducibility and restricts SARS-CoV-2. Mbio 2020, 11, 10–1128. [Google Scholar] [CrossRef]
- Vanderheiden, A.; Ralfs, P.; Chirkova, T.; Upadhyay, A.A.; Zimmerman, M.G.; Bedoya, S.; Aoued, H.; Tharp, G.M.; Pellegrini, K.L.; Manfredi, C.; et al. Type I and Type III Interferons Restrict SARS-CoV-2 Infection of Human Airway Epithelial Cultures. J. Virol. 2020, 94, 10–128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Matuozzo, D.; Le Pen, J.; Lee, D.; Moens, L.; Asano, T.; Bohlen, J.; Liu, Z.; Moncada-Velez, M.; Kendir-Demirkol, Y.; et al. Recessive inborn errors of type I IFN immunity in children with COVID-19 pneumonia. J. Exp. Med. 2022, 219, e20220131. [Google Scholar] [CrossRef] [PubMed]
- Gilli, F. Role of differential expression of interferon receptor isoforms on the response of multiple sclerosis patients to therapy with interferon beta. J. Interf. Cytokine Res. 2010, 30, 733–741. [Google Scholar] [CrossRef] [PubMed]
- de Weerd, N.A.; Matthews, A.Y.; Pattie, P.R.; Bourke, N.M.; Lim, S.S.; Vivian, J.P.; Rossjohn, J.; Hertzog, P.J. A hot spot on interferon α/β receptor subunit 1 (IFNAR1) underpins its interaction with interferon-β and dictates signaling. J. Biol. Chem. 2017, 292, 7554–7565. [Google Scholar] [CrossRef] [PubMed]
- Zoellner, N.; Coesfeld, N.; De Vos, F.H.; Denter, J.; Xu, H.C.; Zimmer, E.; Knebel, B.; Al-Hasani, H.; Mossner, S.; Lang, P.A.; et al. Synthetic mimetics assigned a major role to IFNAR2 in type I interferon signaling. Front. Microbiol. 2022, 13, 947169. [Google Scholar] [CrossRef]


| Inclusion criteria | |
| Population | Patients with COVID-19. |
| Outcome | SNV in IFNAR1 and IFNAR2 is associated with the affectation of an antiviral immune response or poor disease outcome. |
| Types of study design | Observational, case-control, cross-sectional, case reports, and genome-wide association studies (GWAS). |
| Exclusion criteria | |
| Studies on patients who had infections viral infections different from COVID-19. | |
| Studies with incomplete or lacking necessary data. | |
| Studies in pregnant women. | |
| Duplicate studies. | |
| Studies not in the English language. | |
| SNV | Gene Consequence | Minor Allele | MAF (%), 1000 Genomes |
|---|---|---|---|
| rs72550721 | Stop gained | A | 0.70 |
| rs144384060 | Missense variant | A | 0.12 |
| rs749487628 | Missense variant | T | 0.00 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Bielma, M.F.; Falfán-Valencia, R.; Abarca-Rojano, E.; Pérez-Rubio, G. Participation of Single-Nucleotide Variants in IFNAR1 and IFNAR2 in the Immune Response against SARS-CoV-2 Infection: A Systematic Review. Pathogens 2023, 12, 1320. https://doi.org/10.3390/pathogens12111320
López-Bielma MF, Falfán-Valencia R, Abarca-Rojano E, Pérez-Rubio G. Participation of Single-Nucleotide Variants in IFNAR1 and IFNAR2 in the Immune Response against SARS-CoV-2 Infection: A Systematic Review. Pathogens. 2023; 12(11):1320. https://doi.org/10.3390/pathogens12111320
Chicago/Turabian StyleLópez-Bielma, María Fernanda, Ramcés Falfán-Valencia, Edgar Abarca-Rojano, and Gloria Pérez-Rubio. 2023. "Participation of Single-Nucleotide Variants in IFNAR1 and IFNAR2 in the Immune Response against SARS-CoV-2 Infection: A Systematic Review" Pathogens 12, no. 11: 1320. https://doi.org/10.3390/pathogens12111320