
Molecular Characterisation of M. kansasii Isolates by Whole-Genome Sequencing
 ,
,
Abstract
:1. Introduction
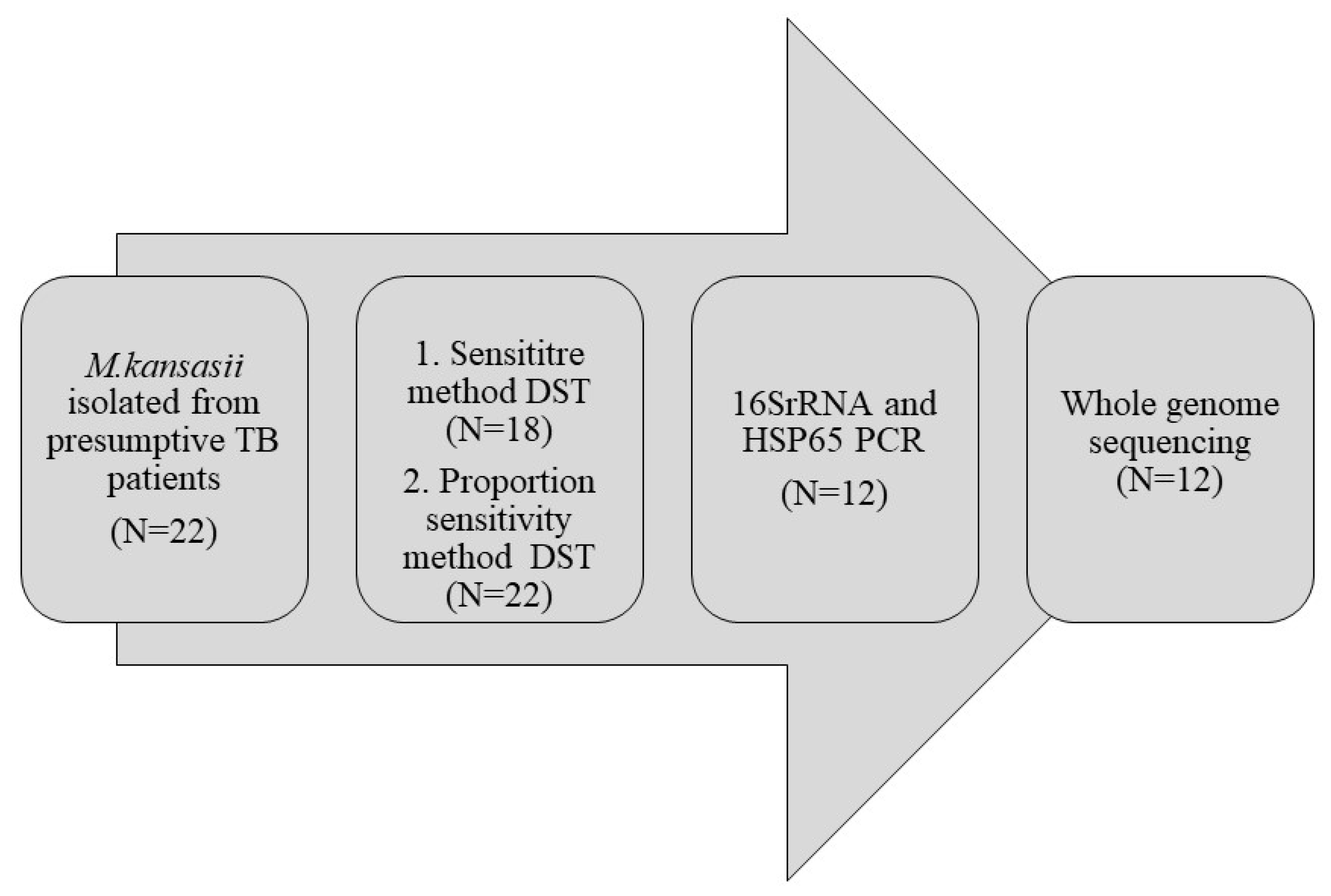
2. Materials and Methods
2.1. Preliminary Genotypic Identification Tests
2.2. Genome Sequencing
2.3. Dataset
2.4. Variant Calling
2.5. Phylogenetic Analysis
2.6. Identification of Mutations in Drug-Resistant Locus
3. Results
3.1. Identification by Molecular Methods
3.2. Subtype Classification
3.3. Association of Mutation with Drug Resistance
3.3.1. RIF and INH Resistance
3.3.2. EMB Resistance
3.3.3. Clarithromycin Resistance
3.3.4. Quinolone Resistance
3.3.5. Amikacin Resistance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tortoli, E. Microbiological features and clinical relevance of new species of the genus Mycobacterium. Clin. Microbiol. Rev. 2014, 27, 727–752. [Google Scholar] [CrossRef]
- Field, S.K.; Cowie, R.L. Lung disease due to the more common nontuberculous mycobacteria. Chest 2006, 129, 1653–1672. [Google Scholar] [CrossRef] [PubMed]
- Prevots, D.R.; Marras, T.K. Epidemiology of human pulmonary infection with nontuberculous mycobacteria: A review. Clin. Chest Med. 2015, 36, 13–34. [Google Scholar] [CrossRef] [PubMed]
- Jagielski, T.; Borowka, P.; Bakula, Z.; Lach, J.; Marciniak, B.; Brzostek, A.; Dziadek, J.; Dziurzynski, M.; Pennings, L.; van Ingen, J.; et al. Genomic Insights Into the Mycobacterium kansasii Complex: An Update. Front. Microbiol. 2019, 10, 2918. [Google Scholar] [CrossRef] [PubMed]
- Tagini, F.; Aeby, S.; Bertelli, C.; Droz, S.; Casanova, C.; Prod’hom, G.; Jaton, K.; Greub, G. Phylogenomics reveal that Mycobacterium kansasii subtypes are species-level lineages. Description of Mycobacterium pseudokansasii sp. nov., Mycobacterium innocens sp. nov. and Mycobacterium attenuatum sp. nov. Int. J. Syst. Evol. Microbiol. 2019, 69, 1696–1704. [Google Scholar] [CrossRef]
- Taillard, C.; Greub, G.; Weber, R.; Pfyffer, G.E.; Bodmer, T.; Zimmerli, S.; Frei, R.; Bassetti, S.; Rohner, P.; Piffaretti, J.C.; et al. Clinical implications of Mycobacterium kansasii species heterogeneity: Swiss National Survey. J. Clin. Microbiol. 2003, 41, 1240–1244. [Google Scholar] [CrossRef]
- Shahraki, A.H.; Trovato, A.; Mirsaeidi, M.; Borroni, E.; Heidarieh, P.; Hashemzadeh, M.; Shahbazi, N.; Cirillo, D.M.; Tortoli, E. Mycobacterium persicum sp. nov., a novel species closely related to Mycobacterium kansasii and Mycobacterium gastri. Int. J. Syst. Evol. Microbiol. 2017, 67, 1766–1770. [Google Scholar] [CrossRef]
- Daley, C.L.; Iaccarino, J.M.; Lange, C.; Cambau, E.; Wallace, R.J., Jr.; Andrejak, C.; Böttger, E.C.; Brozek, J.; Griffith, D.E.; Guglielmetti, L.; et al. Treatment of Nontuberculous Mycobacterial Pulmonary Disease: An Official ATS/ERS/ESCMID/IDSA Clinical Practice Guideline. Clin. Infect. Dis. 2020, 71, e1–e36. [Google Scholar] [CrossRef]
- CLSI. Performance Standards for Susceptibility Testing of Mycobacteia, Nocardia spp., and Other Aerobic Actinonmyces; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2018. [Google Scholar]
- M24-A2; Susceptibility Testing of Mycobacteria, Nocardiae, and Other Aerobic Actinomycetes—2nd Edition. Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2011.
- Rajendran, P.; Padmapriyadarsini, C.; Vijayaraghavan, V.; Manoharan, T.; Lokanathan, L.M.; Kadhar, P.B.; Jayabal, L.; Sivaramakrishnan, G. Drug susceptibility profiling of pulmonary Mycobacterium kansasii and its correlation with treatment outcome. Ann. Thorac. Med. 2021, 16, 323–328. [Google Scholar]
- De Almeida, I.N.; da Silva Carvalho, W.; Rossetti, M.L.; Costa, E.R.D.; De Miranda, S.S. Evaluation of six different DNA extraction methods for detection of Mycobacterium tuberculosis by means of PCR-IS6110: Preliminary study. BMC Res. Notes 2013, 6, 561. [Google Scholar] [CrossRef]
- Mendum, T.A.; Chilima, B.Z.; Hirsch, P.R. The PCR amplification of non-tuberculous mycobacterial 16S rRNA sequences from soil. FEMS Microbiol. Lett. 2000, 185, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Telenti, A.; Marchesi, F.; Balz, M.; Bally, F.; Böttger, E.C.; Bodmer, T. Rapid identification of mycobacteria to the species level by polymerase chain reaction and restriction enzyme analysis. J. Clin. Microbiol. 1993, 31, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Moshiri, N. ViralConsensus: A fast and memory-efficient tool for calling viral consensus genome sequences directly from read alignment data. Bioinformatics 2023, 39, btad317. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Guo, Y.; Cao, Y.; Liu, H.; Yang, J.; Wang, W.; Wang, B.; Li, M.; Yu, F. Clinical and Microbiological Characteristics of Mycobacterium kansasii Pulmonary Infections in China. Microbiol. Spectr. 2022, 10, e0147521. [Google Scholar] [CrossRef]
- World Health Organization. Catalogue of Mutations in Mycobacterium Tuberculosis Complex and Their Association with Drug Resistance; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Walker, T.M.; Miotto, P.; Köser, C.U.; Fowler, P.W.; Knaggs, J.; Iqbal, Z.; Hunt, M.; Chindelevitch, L.; Farhat, M.R.; Cirillo, D.M.; et al. The 2021 WHO catalogue of Mycobacterium tuberculosis complex mutations associated with drug resistance: A genotypic analysis. Lancet Microbe 2022, 3, e265–e273. [Google Scholar] [CrossRef]
- Bakuła, Z.; Modrzejewska, M.; Pennings, L.; Proboszcz, M.; Safianowska, A.; Bielecki, J.; van Ingen, J.; Jagielski, T. Drug Susceptibility Profiling and Genetic Determinants of Drug Resistance in Mycobacterium kansasii. Antimicrob. Agents Chemother. 2018, 62, e01788-17. [Google Scholar] [CrossRef]
- Bikandi, J.; Millán, R.S.; Rementeria, A.; Garaizar, J. In silico analysis of complete bacterial genomes: PCR, AFLP-PCR and endonuclease restriction. Bioinformatics 2004, 20, 798–799. [Google Scholar] [CrossRef] [PubMed]
- Brown-Elliott, B.A.; Wallace, R.J., Jr. Infections caused by nontuberculous mycobacteria other than Mycobacterium avium complex. In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 8th ed.; Bennett, J.E., Dolin, R., Blaser, M.J., Eds.; Elsevier: Philadelphia, PA, USA, 2015. [Google Scholar]
- Rivero-Lezcano, O.M.; Gonzalez-Cortes, C.; Mirsaeidi, M. The unexplained increase of nontuberculous mycobacteriosis. Int. J. Mycobacteriol. 2019, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kalpana, T.; Mugunthan, M.; Joseph, N.M.; Ellappan, K. A Comprehensive Review and Update on Epidemiology, Symptomatology and Management of Nontuberculous Mycobacteria (NTM). J. Pure Appl. Microbiol. 2022, 16, 814–824. [Google Scholar] [CrossRef]
- Sharma, S.K.; Upadhyay, V. Epidemiology, diagnosis & treatment of non-tuberculous mycobacterial diseases. Indian J. Med. Res. 2020, 152, 185–226. [Google Scholar]
- Gomathy, N.S.; Padmapriyadarsini, C.; Silambuchelvi, K.; Nabila, A.; Tamizhselvan, M.; Banurekha, V.V.; Lavanya, J.; Chandrasekar, C. Profile of patients with pulmonary non-tuberculous mycobacterial disease mimicking pulmonary tuberculosis. Indian J. Tuberc. 2019, 66, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Mann, L.B.; Wilson, R.W.; Brown-Elliott, B.A.; Vincent, V.; Iinuma, Y.; Wallace, R.J., Jr. Molecular analysis of Mycobacterium kansasii isolates from the United States. J. Clin. Microbiol. 2004, 42, 119–125. [Google Scholar] [CrossRef]
- Li, Y.; Pang, Y.; Tong, X.; Zheng, H.; Zhao, Y.; Wang, C. Mycobacterium kansasii Subtype I Is Associated with Clarithromycin Resistance in China. Front. Microbiol. 2016, 7, 2097. [Google Scholar] [CrossRef]
- Jagielski, T.; Bakuła, Z.; Roeske, K.; Kamiński, M.; Napiórkowska, A.; Augustynowicz-Kopeć, E.; Zwolska, Z.; Bielecki, J. Detection of mutations associated with isoniazid resistance in multidrug-resistant Mycobacterium tuberculosis clinical isolates. J. Antimicrob. Chemother. 2014, 69, 2369–2375. [Google Scholar] [CrossRef]
- Nasiri, M.J.; Haeili, M.; Ghazi, M.; Goudarzi, H.; Pormohammad, A.; Imani Fooladi, A.A.; Feizabadi, M.M. New Insights in to the Intrinsic and Acquired Drug Resistance Mechanisms in Mycobacteria. Front. Microbiol. 2017, 8, 681. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| S. No. | Lab No. | Resistance to Drugs by Phenotypic DST | Gene Mutations Comparison | Treatment Outcome | Alignment Coverage | |||
|---|---|---|---|---|---|---|---|---|
| MTB Genome | M. kansasii Genome | No. of Reads | Coverage | Mean Depth | ||||
| 1 | NT 08 | RIF/SXT/DOX | embB | No mutation | Died | 5,067,055 | 99.6502 | 38.1 |
| 2 | NT 09 | EMB | embB | No mutation | Cured | 4,350,931 | 99.7226 | 38.2 |
| 3 | NT 12 | CLR/DOX | embB, aftB | aftB | Cured | 4,199,386 | 98.8226 | 37.9 |
| 4 | NT 13 | RIF/SXT/DOX/AMK/LZD/CIP/EMB | embB, aftB | No mutation | Relapse | 4,701,856 | 98.8198 | 38.1 |
| 5 | NT 16 | No resistance | embB, eis | eis | Cured | 4,740,116 | 99.6771 | 37.9 |
| 6 | NT 22 | EMB | embB | embB | Cured | 4,626,353 | 99.3018 | 38 |
| 7 | NT 27 | DOX/EMB | embB | No mutation | Died | 4,862,099 | 99.9468 | 38 |
| 8 | NT 28 | SXT/DOX/ EMB | embB, eis | No mutation | Cured | 4,557,193 | 99.6895 | 38.1 |
| 9 | NT 35 | No resistance | aftB | No mutation | Cured | 3,844,920 | 99.3199 | 38.3 |
| 10 | NT 43 | CLR/RFB/RIF/SXT/AMK/ DOX | embB | No mutation | Cured | 4,094,943 | 99.6353 | 38.1 |
| 11 | NT 33 | No resistance | embB | No mutation | Cured | 4,372,578 | 99.6714 | 38.1 |
| 12 | NT 47 | EMB | No mutation | rrl | Cured | 1,856,345 | 99.5578 | 40.9 |
| S. No. | Strain | Former kansasii Subtype | GenBank No. |
|---|---|---|---|
| 1 | ATCC 12478 | (M. kansasii) I | NC_022663.1_I * |
| 2 | MK7 | (M. kansasii) I | GCA_900565995.1 |
| 3 | Kuro-I | (M. kansasii) I | GCA_014701265.1 |
| 4 | 12MK | (M. persicum) II | NZ_MWQA01000001.1_II * |
| 5 | 1010001469 | (M. persicum) II | LWCM00000000.1 |
| 6 | 3MK | (M. persicum) II | MWKX01.1 |
| 7 | MK142 | (M. pseudokansasii) III | NZ_UPHU01000001.1_III * |
| 8 | 732 | (M. pseudokansasii) III | JANZ01.1 |
| 9 | 174_15_11 | (M. pseudokansasii) III | NKRD01.152 |
| 10 | FDAARGOS_1613 | (M. ostraviense) IV | NZ_CP089224.1_IV * |
| 11 | 241/15 | (M. ostraviense) IV | GCA_002705925.1 |
| 12 | 1010001458 | (M. ostraviense) IV | GCA_001632895.1 |
| 13 | MK21 | (M. innocens) V | NZ_UPHQ01000197.1_V * |
| 14 | 49_11 | (M. innocens) V | NKRC01.1 |
| 15 | 1010001454 | (M. innocens) V | LWCH01.1 |
| 16 | MK41 | (M. attenuatum) VI | NZ_UPHT01000123.1_VI * |
| 17 | MK191 | (M. attenuatum) VI | UPHS01.1 |
| 18 | MK136 | (M. attenuatum) VI | UPHP01.1 |
| Target Drug | Drug-Resistant Locus | Mutation in Comparison to MTB Reference Sequence | Mutation in Comparison to M. kansasii Reference Sequence |
|---|---|---|---|
| Amikacin | eis/MKAN_RS04925 | V301I | M293T |
| E348D | V297I | ||
| D352G | |||
| Ethambutol | embB | S272N | L78M |
| S565G | G130A | ||
| Q853R | A159G | ||
| A1007T | A259T | ||
| Y737N | |||
| aftB | S159A | V100A | |
| I202V | V107A | ||
| S238G | M127V | ||
| R401H | A133V | ||
| M491L | M192L | ||
| V511A | F257L | ||
| A516Q | M331V | ||
| A524G | V339L | ||
| Q561R | I354V | ||
| K399R | |||
| V393L | |||
| L394S | |||
| G412V | |||
| E435D | |||
| T515S | |||
| I541L | |||
| K603R | |||
| S657P |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajendran, P.; Padmapriyadarsini, C.; Nagarajan, N.; Samyuktha, R.; Govindaraju, V.; Golla, R.; Ashokkumar, S.; Shanmugam, S. Molecular Characterisation of M. kansasii Isolates by Whole-Genome Sequencing. Pathogens 2023, 12, 1249. https://doi.org/10.3390/pathogens12101249
Rajendran P, Padmapriyadarsini C, Nagarajan N, Samyuktha R, Govindaraju V, Golla R, Ashokkumar S, Shanmugam S. Molecular Characterisation of M. kansasii Isolates by Whole-Genome Sequencing. Pathogens. 2023; 12(10):1249. https://doi.org/10.3390/pathogens12101249
Chicago/Turabian StyleRajendran, Priya, Chandrasekaran Padmapriyadarsini, Naveenkumar Nagarajan, Roja Samyuktha, Vadivu Govindaraju, Radhika Golla, Shanmugavel Ashokkumar, and Sivakumar Shanmugam. 2023. "Molecular Characterisation of M. kansasii Isolates by Whole-Genome Sequencing" Pathogens 12, no. 10: 1249. https://doi.org/10.3390/pathogens12101249