
Clinical Outcome of Coronavirus Disease 2019 in Patients with Primary Antibody Deficiencies

Abstract
:1. Introduction
2. Primary Antibody Deficiency
3. Origin of SARS-CoV-2
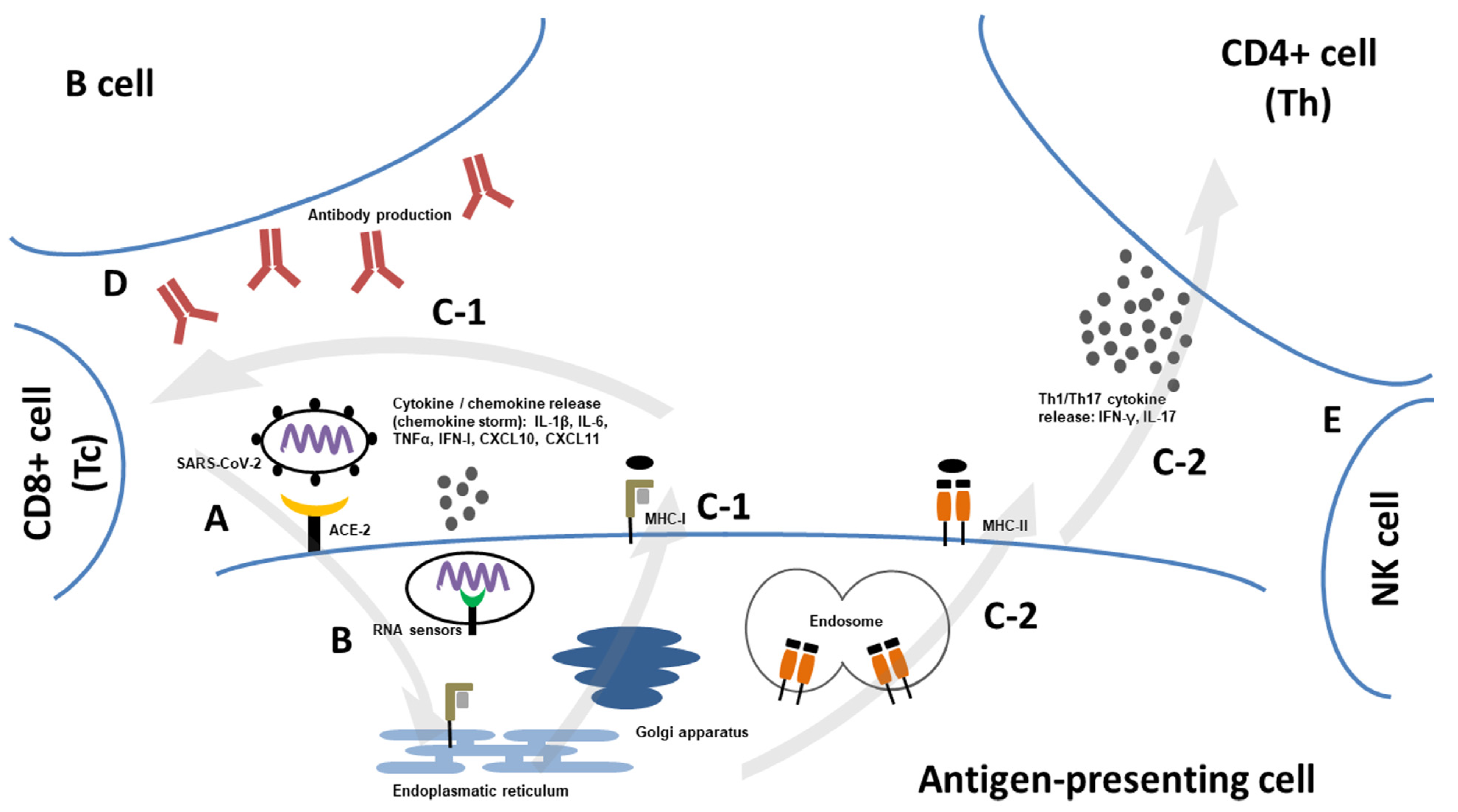
4. Structure and Pathogenesis of SARS-CoV-2
5. COVID-19 in the General Population
6. COVID-19 in Primary Antibody Deficiencies
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. WHO Coronavirus (COVID-19) Dashboard. 2022. Available online: https://covid19.who.int/ (accessed on 24 November 2022).
- Resnick, E.S.; Moshier, E.L.; Godbold, J.H.; Cunningham-Rundles, C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood 2012, 119, 1650–1657. [Google Scholar] [CrossRef]
- Ho, H.-E.; Cunningham-Rundles, C. Non-infectious Complications of Common Variable Immunodeficiency: Updated Clinical Spectrum, Sequelae, and Insights to Pathogenesis. Front. Immunol. 2020, 11, 149. [Google Scholar] [CrossRef] [Green Version]
- Grainger, R.; Kim, A.H.J.; Conway, R.; Yazdany, J.; Robinson, P.C. COVID-19 in people with rheumatic diseases: Risks, outcomes, treatment considerations. Nat. Rev. Rheumatol. 2022, 18, 191–204. [Google Scholar] [CrossRef]
- Belsky, J.A.; Tullius, B.P.; Lamb, M.G.; Sayegh, R.; Stanek, J.R.; Auletta, J.J. COVID-19 in immunocompromised patients: A systematic review of cancer, hematopoietic cell and solid organ transplant patients. J. Infect. 2021, 82, 329–338. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Gasparyan, A.Y.; Ayvazyan, L.; Blackmore, H.; Kitas, G.D. Writing a narrative biomedical review: Considerations for authors, peer reviewers, and editors. Rheumatol. Int. 2011, 31, 1409–1417. [Google Scholar] [CrossRef]
- Abolhassani, H.; Azizi, G.; Sharifi, L.; Yazdani, R.; Mohsenzadegan, M.; Delavari, S.; Sohani, M.; Shirmast, P.; Chavoshzadeh, Z.; Mahdaviani, S.A.; et al. Global systematic review of primary immunodeficiency registries. Expert Rev. Clin. Immunol. 2020, 16, 717–732. [Google Scholar] [CrossRef]
- El-Helou, S.M.; Biegner, A.-K.; Bode, S.; Ehl, S.R.; Heeg, M.; Maccari, M.E.; Ritterbusch, H.; Speckmann, C.; Rusch, S.; Scheible, R.; et al. The German National Registry of Primary Immunodeficiencies (2012–2017). Front. Immunol. 2019, 10, 1272. [Google Scholar] [CrossRef]
- Grimbacher, B. The European Society for Immunodeficiencies (ESID) registry 2014. Clin. Exp. Immunol. 2014, 178 (Suppl. S1), 18–20. [Google Scholar] [CrossRef] [Green Version]
- Bousfiha, A.; Moundir, A.; Tangye, S.G.; Picard, C.; Jeddane, L.; Al-Herz, W.; Rundles, C.C.; Franco, J.L.; Holland, S.M.; Klein, C.; et al. The 2022 Update of IUIS Phenotypical Classification for Human Inborn Errors of Immunity. J. Clin. Immunol. 2022, 42, 1508–1520. [Google Scholar] [CrossRef]
- Yazdani, R.; Azizi, G.; Abolhassani, H.; Aghamohammadi, A. Selective IgA Deficiency: Epidemiology, Pathogenesis, Clinical Phenotype, Diagnosis, Prognosis and Management. Scand. J. Immunol. 2017, 85, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Weifenbach, N.; Schneckenburger, A.A.C.; Lötters, S. Global Distribution of Common Variable Immunodeficiency (CVID) in the Light of the UNDP Human Development Index (HDI): A Preliminary Perspective of a Rare Disease. J. Immunol. Res. 2020, 2020, 8416124. [Google Scholar] [CrossRef]
- Tangye, S.G.; Al-Herz, W.; Bousfiha, A.; Cunningham-Rundles, C.; Franco, J.L.; Holland, S.M.; Klein, C.; Morio, T.; Oksenhendler, E.; Picard, C.; et al. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 2022, 42, 1473–1507. [Google Scholar] [CrossRef]
- Vale, A.M.; Schroeder, H.W. Clinical consequences of defects in B-cell development. J. Allergy Clin. Immunol. 2010, 125, 778–787. [Google Scholar] [CrossRef] [Green Version]
- Cardenas-Morales, M.; Hernandez-Trujillo, V.P. Agammaglobulinemia: From X-linked to Autosomal Forms of Disease. Clin. Rev. Allergy Immunol. 2022, 63, 22–35. [Google Scholar] [CrossRef]
- Conley, M.E.; Broides, A.; Hernandez-Trujillo, V.; Howard, V.; Kanegane, H.; Miyawaki, T.; Shurtleff, S.A. Genetic analysis of patients with defects in early B-cell development. Immunol. Rev. 2005, 203, 216–234. [Google Scholar] [CrossRef]
- Aggarwal, V.; Banday, A.Z.; Jindal, A.K.; Das, J.; Rawat, A. Recent advances in elucidating the genetics of common variable immunodeficiency. Genes Dis. 2020, 7, 26–37. [Google Scholar] [CrossRef]
- Bogaert, D.J.A.; Dullaers, M.; Lambrecht, B.N.; Vermaelen, K.Y.; de Baere, E.; Haerynck, F. Genes associated with common variable immunodeficiency: One diagnosis to rule them all? J. Med. Genet. 2016, 53, 575–590. [Google Scholar] [CrossRef]
- de Valles-Ibáñez, G.; Esteve-Solé, A.; Piquer, M.; González-Navarro, E.A.; Hernandez-Rodriguez, J.; Laayouni, H.; González-Roca, E.; Plaza-Martin, A.M.; Deyà-Martínez, Á.; Martín-Nalda, A.; et al. Evaluating the Genetics of Common Variable Immunodeficiency: Monogenetic Model and Beyond. Front. Immunol. 2018, 9, 636. [Google Scholar] [CrossRef] [Green Version]
- Abolhassani, H.; Aghamohammadi, A.; Hammarström, L. Monogenic mutations associated with IgA deficiency. Expert Rev. Clin. Immunol. 2016, 12, 1321–1335. [Google Scholar] [CrossRef]
- Nechvatalova, J.; Pikulova, Z.; Stikarovska, D.; Pesak, S.; Vlkova, M.; Litzman, J. B-lymphocyte subpopulations in patients with selective IgA deficiency. J. Clin. Immunol. 2012, 32, 441–448. [Google Scholar] [CrossRef]
- Wehr, C.; Kivioja, T.; Schmitt, C.; Ferry, B.; Witte, T.; Eren, E.; Vlkova, M.; Hernandez, M.; Detkova, D.; Bos, P.R.; et al. The EUROclass trial: Defining subgroups in common variable immunodeficiency. Blood 2008, 111, 77–85. [Google Scholar] [CrossRef]
- Warnatz, K.; Schlesier, M. Flowcytometric phenotyping of common variable immunodeficiency. Cytometry B Clin. Cytom. 2008, 74, 261–271. [Google Scholar] [CrossRef]
- Isnardi, I.; Ng, Y.-S.; Menard, L.; Meyers, G.; Saadoun, D.; Srdanovic, I.; Samuels, J.; Berman, J.; Buckner, J.H.; Cunningham-Rundles, C.; et al. Complement receptor 2/CD21- human naive B cells contain mostly autoreactive unresponsive clones. Blood 2010, 115, 5026–5036. [Google Scholar] [CrossRef] [Green Version]
- Unger, S.; Seidl, M.; van Schouwenburg, P.; Rakhmanov, M.; Bulashevska, A.; Frede, N.; Grimbacher, B.; Pfeiffer, J.; Schrenk, K.; Munoz, L.; et al. The TH1 phenotype of follicular helper T cells indicates an IFN-γ-associated immune dysregulation in patients with CD21low common variable immunodeficiency. J. Allergy Clin. Immunol. 2018, 141, 730–740. [Google Scholar] [CrossRef] [Green Version]
- Thorarinsdottir, K.; Camponeschi, A.; Gjertsson, I.; Mårtensson, I.-L. CD21 -/low B cells: A Snapshot of a Unique B Cell Subset in Health and Disease. Scand. J. Immunol. 2015, 82, 254–261. [Google Scholar] [CrossRef]
- Barsotti, N.S.; Almeida, R.R.; Costa, P.R.; Barros, M.T.; Kalil, J.; Kokron, C.M. IL-10-Producing Regulatory B Cells Are Decreased in Patients with Common Variable Immunodeficiency. PLoS ONE 2016, 11, e0151761. [Google Scholar] [CrossRef]
- Azizi, G.; Rezaei, N.; Kiaee, F.; Tavakolinia, N.; Yazdani, R.; Mirshafiey, A.; Aghamohammadi, A. T-Cell Abnormalities in Common Variable Immunodeficiency. J. Investig. Allergol. Clin. Immunol. 2016, 26, 233–243. [Google Scholar] [CrossRef]
- Wong, G.K.; Huissoon, A.P. T-cell abnormalities in common variable immunodeficiency: The hidden defect. J. Clin. Pathol. 2016, 69, 672–676. [Google Scholar] [CrossRef] [Green Version]
- Azizi, G.; Abolhassani, H.; Kiaee, F.; Tavakolinia, N.; Rafiemanesh, H.; Yazdani, R.; Mahdaviani, S.A.; Mohammadikhajehdehi, S.; Tavakol, M.; Ziaee, V.; et al. Autoimmunity and its association with regulatory T cells and B cell subsets in patients with common variable immunodeficiency. Allergol. Immunopathol. (Madr.) 2018, 46, 127–135. [Google Scholar] [CrossRef]
- Malphettes, M.; Gérard, L.; Carmagnat, M.; Mouillot, G.; Vince, N.; Boutboul, D.; Bérezné, A.; Nove-Josserand, R.; Lemoing, V.; Tetu, L.; et al. Late-onset combined immune deficiency: A subset of common variable immunodeficiency with severe T cell defect. Clin. Infect. Dis. 2009, 49, 1329–1338. [Google Scholar] [CrossRef]
- Oksenhendler, E.; Gérard, L.; Fieschi, C.; Malphettes, M.; Mouillot, G.; Jaussaud, R.; Viallard, J.-F.; Gardembas, M.; Galicier, L.; Schleinitz, N.; et al. Infections in 252 patients with common variable immunodeficiency. Clin. Infect. Dis. 2008, 46, 1547–1554. [Google Scholar] [CrossRef]
- Jones, T.P.W.; Buckland, M.; Breuer, J.; Lowe, D.M. Viral infection in primary antibody deficiency syndromes. Rev. Med. Virol. 2019, 29, e2049. [Google Scholar] [CrossRef]
- Janssen, L.M.; van der Flier, M.; de Vries, E. Lessons learned from the clinical presentation of common variable immunodeficiency disorders: A systematic review and meta-analysis. Front. Immunol. 2021, 12, 620709. [Google Scholar] [CrossRef]
- Hanitsch, L.; Baumann, U.; Boztug, K.; Burkhard-Meier, U.; Fasshauer, M.; Habermehl, P.; Hauck, F.; Klock, G.; Liese, J.; Meyer, O.; et al. Treatment and management of primary antibody deficiency: German interdisciplinary evidence-based consensus guideline. Eur. J. Immunol. 2020, 50, 1432–1446. [Google Scholar] [CrossRef]
- Jolles, S.; Orange, J.S.; Gardulf, A.; Stein, M.R.; Shapiro, R.; Borte, M.; Berger, M. Current treatment options with immunoglobulin G for the individualization of care in patients with primary immunodeficiency disease. Clin. Exp. Immunol. 2015, 179, 146–160. [Google Scholar] [CrossRef] [Green Version]
- Abolhassani, H.; Sagvand, B.T.; Shokuhfar, T.; Mirminachi, B.; Rezaei, N.; Aghamohammadi, A. A review on guidelines for management and treatment of common variable immunodeficiency. Expert Rev. Clin. Immunol. 2013, 9, 561–574; quiz 575. [Google Scholar] [CrossRef] [Green Version]
- Cunningham-Rundles, C. How I treat common variable immune deficiency. Blood 2010, 116, 7–15. [Google Scholar] [CrossRef]
- Salzer, U.; Warnatz, K.; Peter, H.H. Common variable immunodeficiency: An update. Arthritis Res. Ther. 2012, 14, 223. [Google Scholar] [CrossRef] [Green Version]
- Mormile, I.; Punziano, A.; Riolo, C.A.; Granata, F.; Williams, M.; de Paulis, A.; Spadaro, G.; Rossi, F.W. Common Variable Immunodeficiency and Autoimmune Diseases: A Retrospective Study of 95 Adult Patients in a Single Tertiary Care Center. Front. Immunol. 2021, 12, 652487. [Google Scholar] [CrossRef]
- Lo, B.; Zhang, K.; Lu, W.; Zheng, L.; Zhang, Q.; Kanellopoulou, C.; Zhang, Y.; Liu, Z.; Fritz, J.M.; Marsh, R.; et al. AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science 2015, 349, 436–440. [Google Scholar] [CrossRef] [Green Version]
- Egg, D.; Rump, I.C.; Mitsuiki, N.; Rojas-Restrepo, J.; Maccari, M.-E.; Schwab, C.; Gabrysch, A.; Warnatz, K.; Goldacker, S.; Patiño, V.; et al. Therapeutic options for CTLA-4 insufficiency. J. Allergy Clin. Immunol. 2022, 149, 736–746. [Google Scholar] [CrossRef]
- Coulter, T.I.; Cant, A.J. The Treatment of Activated PI3Kδ Syndrome. Front. Immunol. 2018, 9, 2043. [Google Scholar] [CrossRef] [Green Version]
- Maccari, M.E.; Abolhassani, H.; Aghamohammadi, A.; Aiuti, A.; Aleinikova, O.; Bangs, C.; Baris, S.; Barzaghi, F.; Baxendale, H.; Buckland, M.; et al. Disease Evolution and Response to Rapamycin in Activated Phosphoinositide 3-Kinase δ Syndrome: The European Society for Immunodeficiencies-Activated Phosphoinositide 3-Kinase δ Syndrome Registry. Front. Immunol. 2018, 9, 543. [Google Scholar] [CrossRef] [Green Version]
- Pecoraro, A.; Crescenzi, L.; Galdiero, M.R.; Marone, G.; Rivellese, F.; Rossi, F.W.; de Paulis, A.; Genovese, A.; Spadaro, G. Immunosuppressive therapy with rituximab in common variable immunodeficiency. Clin. Mol. Allergy 2019, 17, 9. [Google Scholar] [CrossRef] [Green Version]
- de Wit, E.; van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef]
- Holmes, E.C.; Goldstein, S.A.; Rasmussen, A.L.; Robertson, D.L.; Crits-Christoph, A.; Wertheim, J.O.; Anthony, S.J.; Barclay, W.S.; Boni, M.F.; Doherty, P.C.; et al. The origins of SARS-CoV-2: A critical review. Cell 2021, 184, 4848–4856. [Google Scholar] [CrossRef]
- Prince, T.; Smith, S.L.; Radford, A.D.; Solomon, T.; Hughes, G.L.; Patterson, E.I. SARS-CoV-2 Infections in Animals: Reservoirs for Reverse Zoonosis and Models for Study. Viruses 2021, 13, 494. [Google Scholar] [CrossRef]
- Yang, H.; Rao, Z. Structural biology of SARS-CoV-2 and implications for therapeutic development. Nat. Rev. Microbiol. 2021, 19, 685–700. [Google Scholar] [CrossRef]
- Wang, M.-Y.; Zhao, R.; Gao, L.-J.; Gao, X.-F.; Wang, D.-P.; Cao, J.-M. SARS-CoV-2, Structure, Biology, and Structure-Based Therapeutics Development. Front. Cell Infect. Microbiol. 2020, 10, 587269. [Google Scholar] [CrossRef]
- Sridhar, S.; Nicholls, J. Pathophysiology of infection with SARS-CoV-2-What is known and what remains a mystery. Respirology 2021, 26, 652–665. [Google Scholar] [CrossRef]
- Cevik, M.; Kuppalli, K.; Kindrachuk, J.; Peiris, M. Virology, transmission, and pathogenesis of SARS-CoV-2. BMJ 2020, 371, m3862. [Google Scholar] [CrossRef]
- Chu, H.; Yuen, K.-Y. Pathogenicity of SARS-CoV-2 Omicron. Clin. Transl. Med. 2022, 12, e880. [Google Scholar] [CrossRef]
- Diamond, M.S.; Kanneganti, T.-D. Innate immunity: The first line of defense against SARS-CoV-2. Nat. Immunol. 2022, 23, 165–176. [Google Scholar] [CrossRef]
- Lamers, M.M.; Haagmans, B.L. SARS-CoV-2 pathogenesis. Nat. Rev. Microbiol. 2022, 20, 270–284. [Google Scholar] [CrossRef]
- Del Valle, D.M.; Kim-Schulze, S.; Huang, H.-H.; Beckmann, N.D.; Nirenberg, S.; Wang, B.; Lavin, Y.; Swartz, T.H.; Madduri, D.; Stock, A.; et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020, 26, 1636–1643. [Google Scholar] [CrossRef]
- Azkur, A.K.; Akdis, M.; Azkur, D.; Sokolowska, M.; van de Veen, W.; Brüggen, M.-C.; O’Mahony, L.; Gao, Y.; Nadeau, K.; Akdis, C.A. Immune response to SARS-CoV-2 and mechanisms of immunopathological changes in COVID-19. Allergy 2020, 75, 1564–1581. [Google Scholar] [CrossRef]
- Gusev, E.; Sarapultsev, A.; Solomatina, L.; Chereshnev, V. SARS-CoV-2-Specific Immune Response and the Pathogenesis of COVID-19. Int. J. Mol. Sci. 2022, 23, 1716. [Google Scholar] [CrossRef]
- Yaugel-Novoa, M.; Bourlet, T.; Paul, S. Role of the humoral immune response during COVID-19, guilty or not guilty? Mucosal Immunol. 2022, 15, 1170–1180. [Google Scholar] [CrossRef]
- Milota, T.; Strizova, Z.; Smetanova, J.; Sediva, A. An immunologist’s perspective on anti-COVID-19 vaccines. Curr. Opin. Allergy Clin. Immunol. 2021, 21, 545–552. [Google Scholar] [CrossRef]
- Qi, H.; Liu, B.; Wang, X.; Zhang, L. The humoral response and antibodies against SARS-CoV-2 infection. Nat. Immunol. 2022, 23, 1008–1020. [Google Scholar] [CrossRef]
- Moss, P. The T cell immune response against SARS-CoV-2. Nat. Immunol. 2022, 23, 186–193. [Google Scholar] [CrossRef]
- Niessl, J.; Sekine, T.; Buggert, M. T cell immunity to SARS-CoV-2. Semin. Immunol. 2021, 55, 101505. [Google Scholar] [CrossRef]
- Vardhana, S.; Baldo, L.; Morice, W.G.; Wherry, E.J. Understanding T cell responses to COVID-19 is essential for informing public health strategies. Sci. Immunol. 2022, 7, eabo1303. [Google Scholar] [CrossRef]
- Geers, D.; Shamier, M.C.; Bogers, S.; den Hartog, G.; Gommers, L.; Nieuwkoop, N.N.; Schmitz, K.S.; Rijsbergen, L.C.; van Osch, J.A.T.; Dijkhuizen, E.; et al. SARS-CoV-2 variants of concern partially escape humoral but not T-cell responses in COVID-19 convalescent donors and vaccinees. Sci. Immunol. 2021, 6, abj1750. [Google Scholar] [CrossRef]
- Mlcochova, P.; Kemp, S.A.; Dhar, M.S.; Papa, G.; Meng, B.; Ferreira, I.A.T.M.; Datir, R.; Collier, D.A.; Albecka, A.; Singh, S.; et al. SARS-CoV-2 B.1.617.2 Delta variant replication and immune evasion. Nature 2021, 599, 114–119. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Fan, Y.; Li, X.; Zhang, L.; Wan, S.; Zhang, L.; Zhou, F. SARS-CoV-2 Omicron variant: Recent progress and future perspectives. Signal Transduct. Target. Ther. 2022, 7, 141. [Google Scholar] [CrossRef]
- Li, Q.; Wang, Y.; Sun, Q.; Knopf, J.; Herrmann, M.; Lin, L.; Jiang, J.; Shao, C.; Li, P.; He, X.; et al. Immune response in COVID-19: What is next? Cell Death Differ. 2022, 29, 1107–1122. [Google Scholar] [CrossRef]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Adhikari, S.P.; Meng, S.; Wu, Y.-J.; Mao, Y.-P.; Ye, R.-X.; Wang, Q.-Z.; Sun, C.; Sylvia, S.; Rozelle, S.; Raat, H.; et al. Epidemiology, causes, clinical manifestation and diagnosis, prevention and control of coronavirus disease (COVID-19) during the early outbreak period: A scoping review. Infect. Dis. Poverty 2020, 9, 29. [Google Scholar] [CrossRef] [Green Version]
- Vetter, P.; Vu, D.L.; L’Huillier, A.G.; Schibler, M.; Kaiser, L.; Jacquerioz, F. Clinical features of COVID-19. BMJ 2020, 369, m1470. [Google Scholar] [CrossRef] [Green Version]
- Da Rosa Mesquita, R.; Francelino Silva Junior, L.C.; Santos Santana, F.M.; Farias de Oliveira, T.; Campos Alcântara, R.; Monteiro Arnozo, G.; Da Rodrigues Silva Filho, E.; Galdino Dos Santos, A.G.; Da Oliveira Cunha, E.J.; Salgueiro de Aquino, S.H.; et al. Clinical manifestations of COVID-19 in the general population: Systematic review. Wien. Klin. Wochenschr. 2021, 133, 377–382. [Google Scholar] [CrossRef]
- Mendiola-Pastrana, I.R.; López-Ortiz, E.; La Río de Loza-Zamora, J.G.; González, J.; Gómez-García, A.; López-Ortiz, G. SARS-CoV-2 Variants and Clinical Outcomes: A Systematic Review. Life 2022, 12, 170. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.W.X.; Chiew, C.J.; Ang, L.W.; Mak, T.M.; Cui, L.; Toh, M.P.H.S.; Lim, Y.D.; Lee, P.H.; Lee, T.H.; Chia, P.Y.; et al. Clinical and Virological Features of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Variants of Concern: A Retrospective Cohort Study Comparing B.1.1.7 (Alpha), B.1.351 (Beta), and B.1.617.2 (Delta). Clin. Infect. Dis. 2022, 75, e1128–e1136. [Google Scholar] [CrossRef] [PubMed]
- Zali, A.; Khodadoost, M.; Gholamzadeh, S.; Janbazi, S.; Piri, H.; Taraghikhah, N.; Hannani, K.; Looha, M.A.; Mohammadi, G. Mortality among hospitalized COVID-19 patients during surges of SARS-CoV-2 alpha (B.1.1.7) and delta (B.1.617.2) variants. Sci. Rep. 2022, 12, 18918. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Quadri, S.; AlAwadhi, A.I.; AlQahtani, M. COVID-19 Recovery Patterns Across Alpha (B.1.1.7) and Delta (B.1.617.2) Variants of SARS-CoV-2. Front. Immunol. 2022, 13, 812606. [Google Scholar] [CrossRef]
- Esper, F.P.; Adhikari, T.M.; Tu, Z.J.; Cheng, Y.W.; El-Haddad, K.; Farkas, D.H.; Bosler, D.; Rhoads, D.; Procop, G.W.; Ko, J.S.; et al. Alpha to Omicron: Disease Severity and Clinical Outcomes of Major SARS-CoV-2 Variants. J. Infect. Dis. 2022. [Google Scholar] [CrossRef] [PubMed]
- Lewnard, J.A.; Hong, V.X.; Patel, M.M.; Kahn, R.; Lipsitch, M.; Tartof, S.Y. Clinical outcomes associated with SARS-CoV-2 Omicron (B.1.1.529) variant and BA.1/BA.1.1 or BA.2 subvariant infection in Southern California. Nat. Med. 2022, 28, 1933–1943. [Google Scholar] [CrossRef] [PubMed]
- Skarbinski, J.; Wood, M.S.; Chervo, T.C.; Schapiro, J.M.; Elkin, E.P.; Valice, E.; Amsden, L.B.; Hsiao, C.; Quesenberry, C.; Corley, D.A.; et al. Risk of severe clinical outcomes among persons with SARS-CoV-2 infection with differing levels of vaccination during widespread Omicron (B.1.1.529) and Delta (B.1.617.2) variant circulation in Northern California: A retrospective cohort study. Lancet Reg. Health Am. 2022, 12, 100297. [Google Scholar] [CrossRef] [PubMed]
- Ward, I.L.; Bermingham, C.; Ayoubkhani, D.; Gethings, O.J.; Pouwels, K.B.; Yates, T.; Khunti, K.; Hippisley-Cox, J.; Banerjee, A.; Walker, A.S.; et al. Risk of COVID-19 related deaths for SARS-CoV-2 omicron (B.1.1.529) compared with delta (B.1.617.2): Retrospective cohort study. BMJ 2022, 378, e070695. [Google Scholar] [CrossRef] [PubMed]
- Bálint, G.; Vörös-Horváth, B.; Széchenyi, A. Omicron: Increased transmissibility and decreased pathogenicity. Signal Transduct. Target Ther. 2022, 7, 151. [Google Scholar] [CrossRef]
- Michlmayr, D.; Hansen, C.H.; Gubbels, S.M.; Valentiner-Branth, P.; Bager, P.; Obel, N.; Drewes, B.; Møller, C.H.; Møller, F.T.; Legarth, R.; et al. Observed protection against SARS-CoV-2 reinfection following a primary infection: A Danish cohort study among unvaccinated using two years of nationwide PCR-test data. Lancet Reg. Health Eur. 2022, 20, 100452. [Google Scholar] [CrossRef] [PubMed]
- Estimating excess mortality due to the COVID-19 pandemic: A systematic analysis of COVID-19-related mortality, 2020–2021. Lancet 2022, 399, 1513–1536. [CrossRef] [PubMed]
- Schöley, J.; Aburto, J.M.; Kashnitsky, I.; Kniffka, M.S.; Zhang, L.; Jaadla, H.; Dowd, J.B.; Kashyap, R. Life expectancy changes since COVID-19. Nat. Hum. Behav. 2022, 6, 1649–1659. [Google Scholar] [CrossRef]
- Booth, A.; Reed, A.B.; Ponzo, S.; Yassaee, A.; Aral, M.; Plans, D.; Labrique, A.; Mohan, D. Population risk factors for severe disease and mortality in COVID-19: A global systematic review and meta-analysis. PLoS One 2021, 16, e0247461. [Google Scholar] [CrossRef]
- Schröder, J.; Kahlke, V.; Staubach, K.H.; Zabel, P.; Stüber, F. Gender differences in human sepsis. Arch. Surg. 1998, 133, 1200–1205. [Google Scholar] [CrossRef]
- Klein, S.L.; Flanagan, K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef]
- Peckham, H.; de Gruijter, N.M.; Raine, C.; Radziszewska, A.; Ciurtin, C.; Wedderburn, L.R.; Rosser, E.C.; Webb, K.; Deakin, C.T. Male sex identified by global COVID-19 meta-analysis as a risk factor for death and Itu Admission. Nat. Commun. 2020, 11, 6317. [Google Scholar] [CrossRef]
- Bienvenu, L.A.; Noonan, J.; Wang, X.; Peter, K. Higher mortality of COVID-19 in males: Sex differences in immune response and cardiovascular comorbidities. Cardiovasc. Res. 2020, 116, 2197–2206. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.S.; Moulin, T.C.; Schiöth, H.B. Sex differences in COVID-19, the role of androgens in disease severity and progression. Endocr 2021, 71, 3–8. [Google Scholar] [CrossRef]
- Sunjaya, A.P.; Allida, S.M.; Di Tanna, G.L.; Jenkins, C.R. Asthma and COVID-19 risk: A systematic review and meta-analysis. Eur. Respir. J. 2022, 59, 2101209. [Google Scholar] [CrossRef]
- Griesel, M.; Wagner, C.; Mikolajewska, A.; Stegemann, M.; Fichtner, F.; Metzendorf, M.-I.; Nair, A.A.; Daniel, J.; Fischer, A.-L.; Skoetz, N. Inhaled corticosteroids for the treatment of COVID-19. Cochrane Database Syst. Rev. 2022, 3, CD015125. [Google Scholar] [CrossRef]
- Choi, J.H.; Choi, S.-H.; Yun, K.W. Risk Factors for Severe COVID-19 in Children: A Systematic Review and Meta-Analysis. J. Korean Med. Sci. 2022, 37, e35. [Google Scholar] [CrossRef]
- Castanares-Zapatero, D.; Chalon, P.; Kohn, L.; Dauvrin, M.; Detollenaere, J.; Maertens de Noordhout, C.; Primus-de Jong, C.; Cleemput, I.; van den Heede, K. Pathophysiology and mechanism of long COVID: A comprehensive review. Ann. Med. 2022, 54, 1473–1487. [Google Scholar] [CrossRef]
- Mantovani, A.; Morrone, M.C.; Patrono, C.; Santoro, M.G.; Schiaffino, S.; Remuzzi, G.; Bussolati, G. Long Covid: Where we stand and challenges ahead. Cell Death Differ. 2022, 29, 1891–1900. [Google Scholar] [CrossRef]
- Michelen, M.; Manoharan, L.; Elkheir, N.; Cheng, V.; Dagens, A.; Hastie, C.; O’Hara, M.; Suett, J.; Dahmash, D.; Bugaeva, P.; et al. Characterising long COVID: A living systematic review. BMJ Glob. Health 2021, 6, e005427. [Google Scholar] [CrossRef] [PubMed]
- Crook, H.; Raza, S.; Nowell, J.; Young, M.; Edison, P. Long covid-mechanisms, risk factors, and management. BMJ 2021, 374, n1648. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Nirantharakumar, K.; Hughes, S.; Myles, P.; Williams, T.; Gokhale, K.M.; Taverner, T.; Chandan, J.S.; Brown, K.; Simms-Williams, N.; et al. Symptoms and risk factors for long COVID in non-hospitalized adults. Nat. Med. 2022, 28, 1706–1714. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Tomasoni, D.; Falcinella, C.; Barbanotti, D.; Castoldi, R.; Mulè, G.; Augello, M.; Mondatore, D.; Allegrini, M.; Cona, A.; et al. Female gender is associated with long COVID syndrome: A prospective cohort study. Clin. Microbiol. Infect. 2022, 28, e9–e611. [Google Scholar] [CrossRef]
- Marcus, N.; Frizinsky, S.; Hagin, D.; Ovadia, A.; Hanna, S.; Farkash, M.; Maoz-Segal, R.; Agmon-Levin, N.; Broides, A.; Nahum, A.; et al. Minor Clinical Impact of COVID-19 Pandemic on Patients With Primary Immunodeficiency in Israel. Front. Immunol. 2020, 11, 614086. [Google Scholar] [CrossRef]
- Goudouris, E.S.; Pinto-Mariz, F.; Mendonça, L.O.; Aranda, C.S.; Guimarães, R.R.; Kokron, C.; Barros, M.T.; Anísio, F.; Alonso, M.L.O.; Marcelino, F.; et al. Outcome of SARS-CoV-2 Infection in 121 Patients with Inborn Errors of Immunity: A Cross-Sectional Study. J. Clin. Immunol. 2021, 41, 1479–1489. [Google Scholar] [CrossRef]
- Meyts, I.; Bucciol, G.; Quinti, I.; Neven, B.; Fischer, A.; Seoane, E.; Lopez-Granados, E.; Gianelli, C.; Robles-Marhuenda, A.; Jeandel, P.-Y.; et al. Coronavirus disease 2019 in patients with inborn errors of immunity: An international study. J. Allergy Clin. Immunol. 2021, 147, 520–531. [Google Scholar] [CrossRef]
- Shields, A.M.; Burns, S.O.; Savic, S.; Richter, A.G. COVID-19 in patients with primary and secondary immunodeficiency: The United Kingdom experience. J. Allergy Clin. Immunol. 2021, 147, 870–875.e1. [Google Scholar] [CrossRef]
- Milota, T.; Sobotkova, M.; Smetanova, J.; Bloomfield, M.; Vydlakova, J.; Chovancova, Z.; Litzman, J.; Hakl, R.; Novak, J.; Malkusova, I.; et al. Risk Factors for Severe COVID-19 and Hospital Admission in Patients With Inborn Errors of Immunity-Results From a Multicenter Nationwide Study. Front. Immunol. 2022, 13, 835770. [Google Scholar] [CrossRef]
- Steiner, S.; Schwarz, T.; Corman, V.M.; Gebert, L.; Kleinschmidt, M.C.; Wald, A.; Gläser, S.; Kruse, J.M.; Zickler, D.; Peric, A.; et al. SARS-CoV-2 T Cell Response in Severe and Fatal COVID-19 in Primary Antibody Deficiency Patients Without Specific Humoral Immunity. Front. Immunol. 2022, 13, 840126. [Google Scholar] [CrossRef]
- Milito, C.; Lougaris, V.; Giardino, G.; Punziano, A.; Vultaggio, A.; Carrabba, M.; Cinetto, F.; Scarpa, R.; Delle Piane, R.M.; Baselli, L.; et al. Clinical outcome, incidence, and SARS-CoV-2 infection-fatality rates in Italian patients with inborn errors of immunity. J. Allergy Clin. Immunol. Pract. 2021, 9, 2904–2906.e2. [Google Scholar] [CrossRef]
- Castano-Jaramillo, L.M.; Yamazaki-Nakashimada, M.A.; O’Farrill-Romanillos, P.M.; Muzquiz Zermeño, D.; Scheffler Mendoza, S.C.; Venegas Montoya, E.; García Campos, J.A.; Sánchez-Sánchez, L.M.; Gámez González, L.B.; Ramírez López, J.M.; et al. COVID-19 in the Context of Inborn Errors of Immunity: A Case Series of 31 Patients from Mexico. J. Clin. Immunol. 2021, 41, 1463–1478. [Google Scholar] [CrossRef] [PubMed]
- Karakoc Aydiner, E.; Bilgic Eltan, S.; Babayeva, R.; Aydiner, O.; Kepenekli, E.; Kolukisa, B.; Sefer, A.P.; Yalcin Gungoren, E.; Karabiber, E.; Yucel, E.O.; et al. Adverse COVID-19 outcomes in immune deficiencies: Inequality exists between subclasses. Allergy 2022, 77, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Esenboga, S.; Ocak, M.; Akarsu, A.; Bildik, H.N.; Cagdas, D.; Iskit, A.T.; Tezcan, I. COVID-19 in Patients with Primary Immunodeficiency. J. Clin. Immunol. 2021, 41, 1515–1522. [Google Scholar] [CrossRef] [PubMed]
- Kołtan, S.; Ziętkiewicz, M.; Grześk, E.; Becht, R.; Berdej-Szczot, E.; Cienkusz, M.; Ewertowska, M.; Heropolitańska-Pliszka, E.; Krysiak, N.; Lewandowicz-Uszyńska, A.; et al. COVID-19 in unvaccinated patients with inborn errors of immunity-polish experience. Front. Immunol. 2022, 13, 953700. [Google Scholar] [CrossRef]
- Ho, H.-E.; Mathew, S.; Peluso, M.J.; Cunningham-Rundles, C. Clinical outcomes and features of COVID-19 in patients with primary immunodeficiencies in New York City. J. Allergy Clin. Immunol. Pract. 2021, 9, 490–493.e2. [Google Scholar] [CrossRef]
- Moazzen, N.; Ahanchian, H.; Aelami, M.H.; Asiyon, H.; Astaneh, M.; Naeimi, A.M.; Rezaei, N. COVID-19 in children with inborn errors of immunity: Clinical scenarios. Am. J. Clin. Exp. Immunol. 2021, 10, 77–85. [Google Scholar]
- Delmonte, O.M.; Castagnoli, R.; Notarangelo, L.D. COVID-19 and inborn errors of immunity. Physiology 2022, 37, 290–301. [Google Scholar] [CrossRef]
- Abolhassani, H.; Delavari, S.; Landegren, N.; Shokri, S.; Bastard, P.; Du, L.; Zuo, F.; Hajebi, R.; Abolnezhadian, F.; Iranparast, S.; et al. Genetic and immunologic evaluation of children with inborn errors of immunity and severe or critical COVID-19. J. Allergy Clin. Immunol. 2022, 150, 1059–1073. [Google Scholar] [CrossRef]
- National Institutes of Health. Coronavirus Disease 2019 (COVID-19) Treatment Guidelines. 10 November 2022. Available online: https://www.covid19treatmentguidelines.nih.gov/ (accessed on 24 November 2022).



{kind=link}
{kind=link}
| Manifestation | Pooled Proportion (%, 95%CI) |
|---|---|
| Chronic lung disease | 44 [27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63] |
| Lymphadenopathy | 30 (20–42) |
| Splenomegaly | 29 (22–37) |
| Autoimmunity | 27 (22–32) |
| Hepatomegaly | 14 (8–22) |
| Enteropathy | 9 (6–13) |
| Lymphoid malignancy | 5 (3–7) |
| Gastric cancer | 2 (1–4) |
| Risk Factor | Severe (OR, 95%CI) | Mortality (OR, 95%CI) |
|---|---|---|
| Cardiovascular disease | 3.37 (2.89–3.85) | 4.04 (1.95–6.13) |
| COPD | 2.47 (1.44–3.51) | 2.68 (1.8–3.55) |
| CKD | 3.5 (1.4–5.59) | 2.79 (1.19–4.4) |
| Obesity | 2.02 (1.02–3.01) | N/A |
| Higher age (>65) | 2.14 (1.0–3.29) | 1.89 (0.09–3.69) |
| Active smoking | 1.22 (0.87–1.57) | 2.13 (2.08–2.12) |
| Immunosuppression | 1.17 (0.96–1.38) | 2.31 (1.96–2.65) |
| Male sex | 1.62 (1.29–1.94) | 1.94 (1.51–2.37) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milota, T.; Smetanova, J.; Bartunkova, J. Clinical Outcome of Coronavirus Disease 2019 in Patients with Primary Antibody Deficiencies. Pathogens 2023, 12, 109. https://doi.org/10.3390/pathogens12010109
Milota T, Smetanova J, Bartunkova J. Clinical Outcome of Coronavirus Disease 2019 in Patients with Primary Antibody Deficiencies. Pathogens. 2023; 12(1):109. https://doi.org/10.3390/pathogens12010109
Chicago/Turabian StyleMilota, Tomas, Jitka Smetanova, and Jirina Bartunkova. 2023. "Clinical Outcome of Coronavirus Disease 2019 in Patients with Primary Antibody Deficiencies" Pathogens 12, no. 1: 109. https://doi.org/10.3390/pathogens12010109