
Embedding of HIV Egress within Cortical F-Actin

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
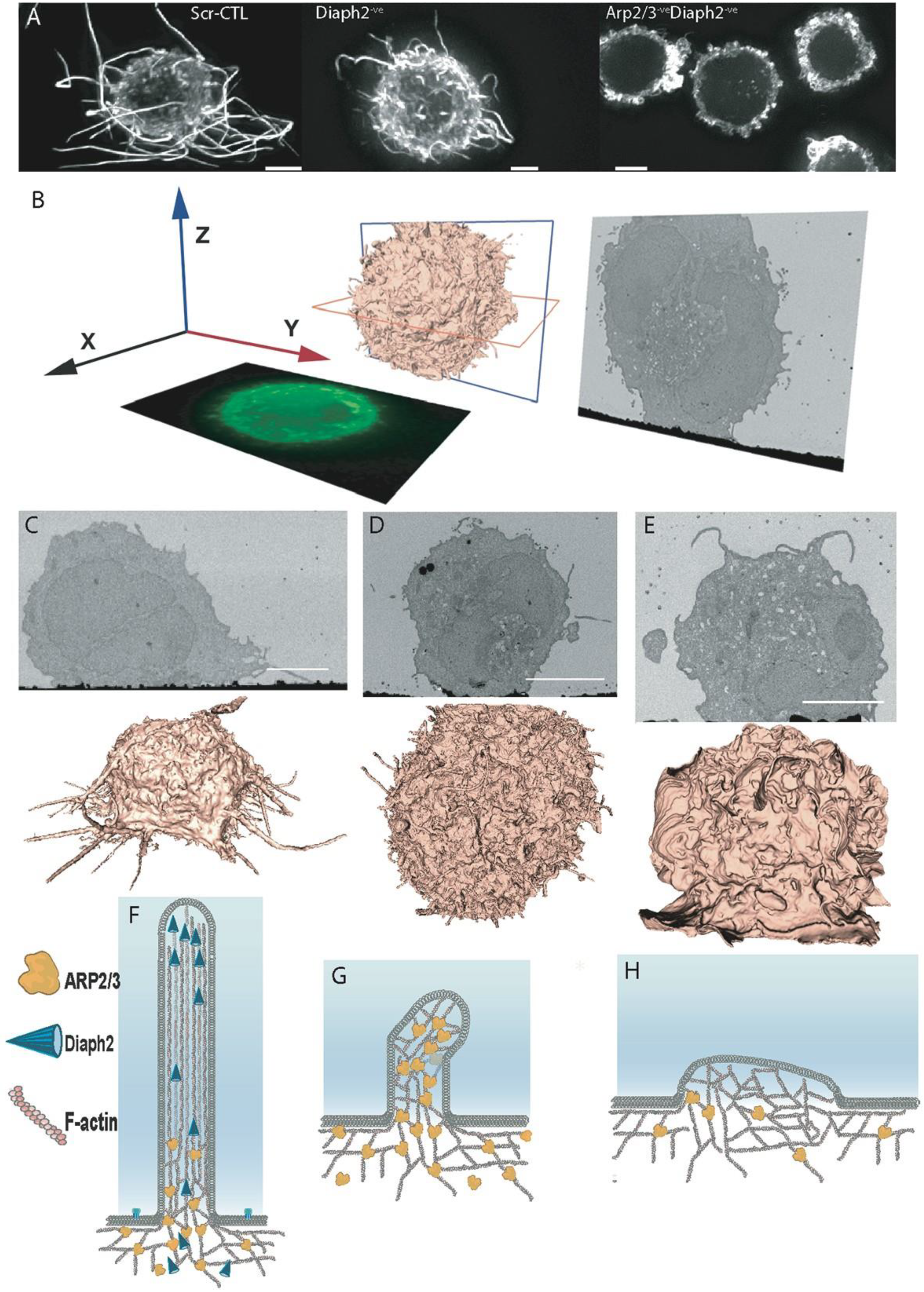
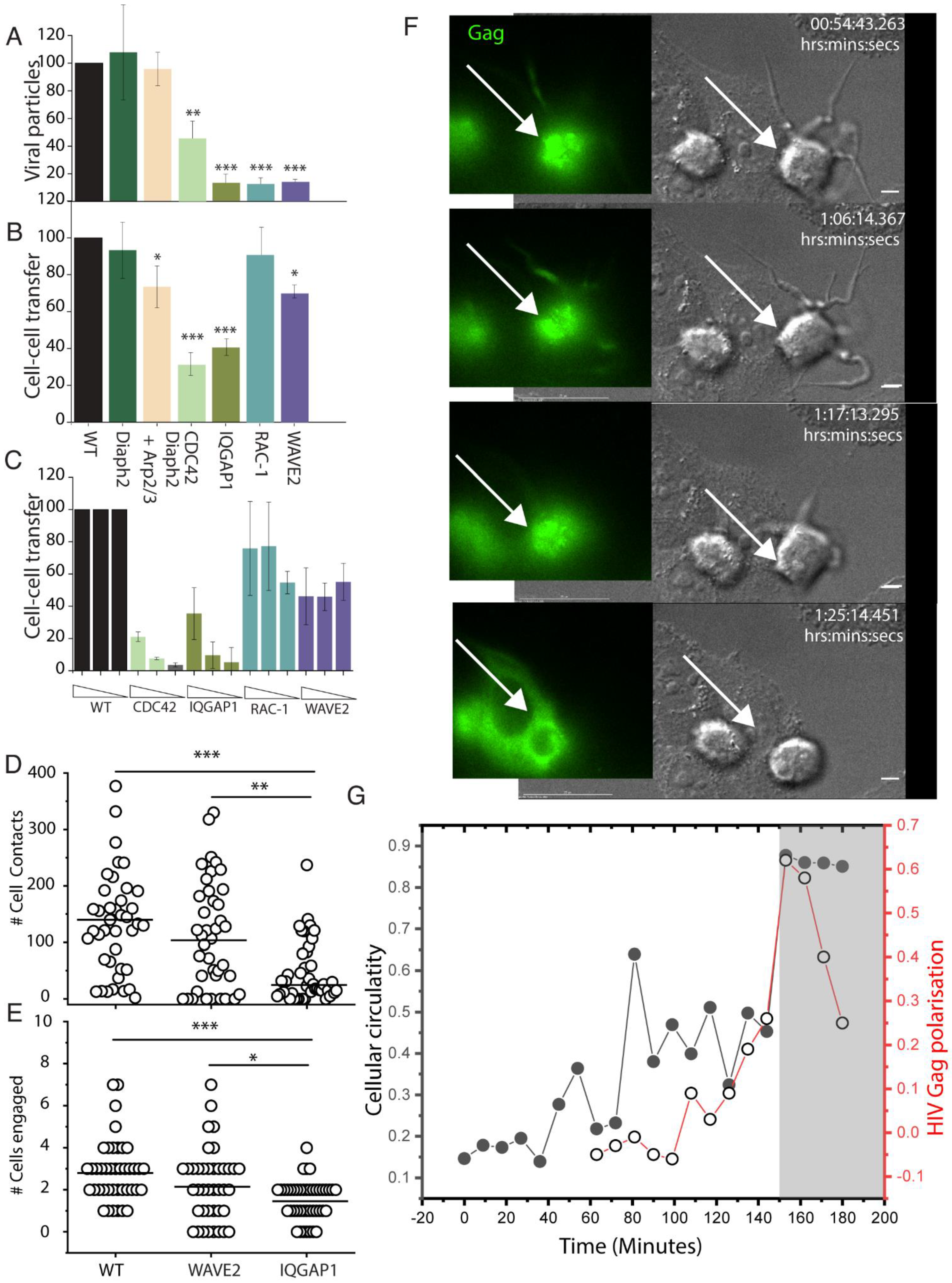
2.1. Moulding Cortical F-Actin through Formin and Arp2/3 Depletion
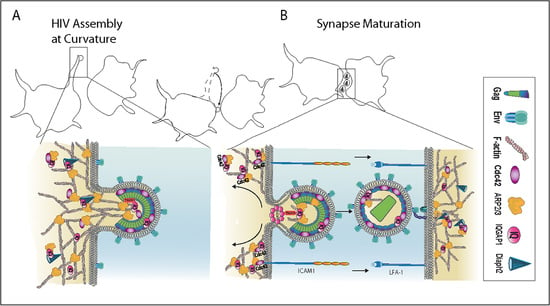
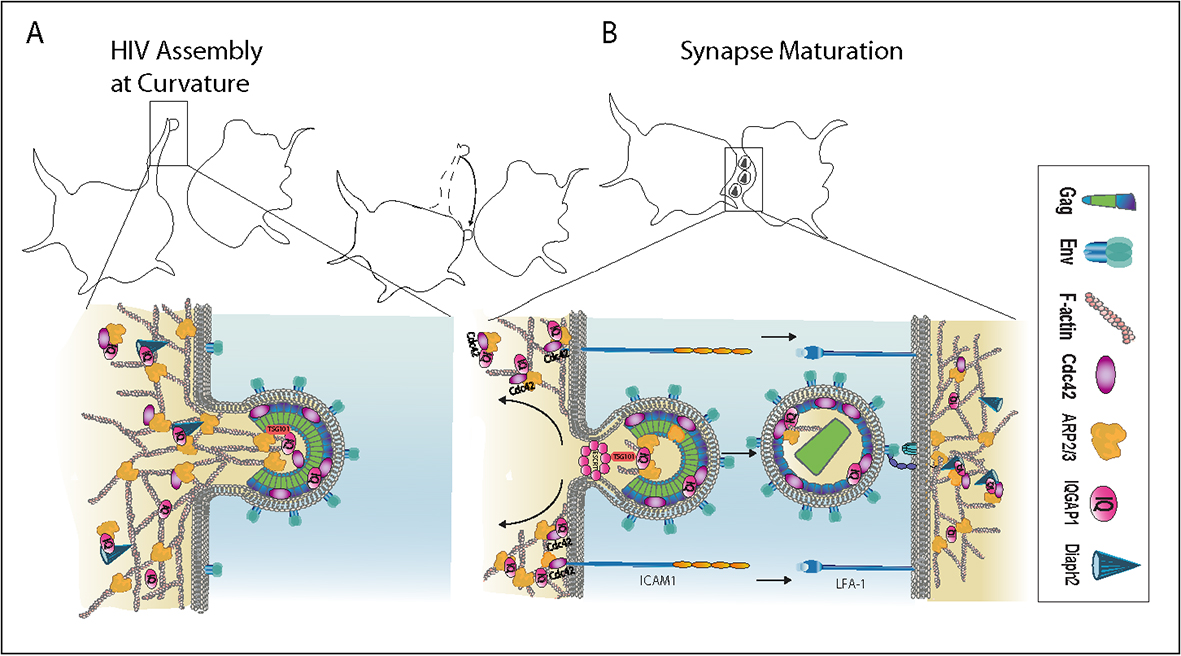
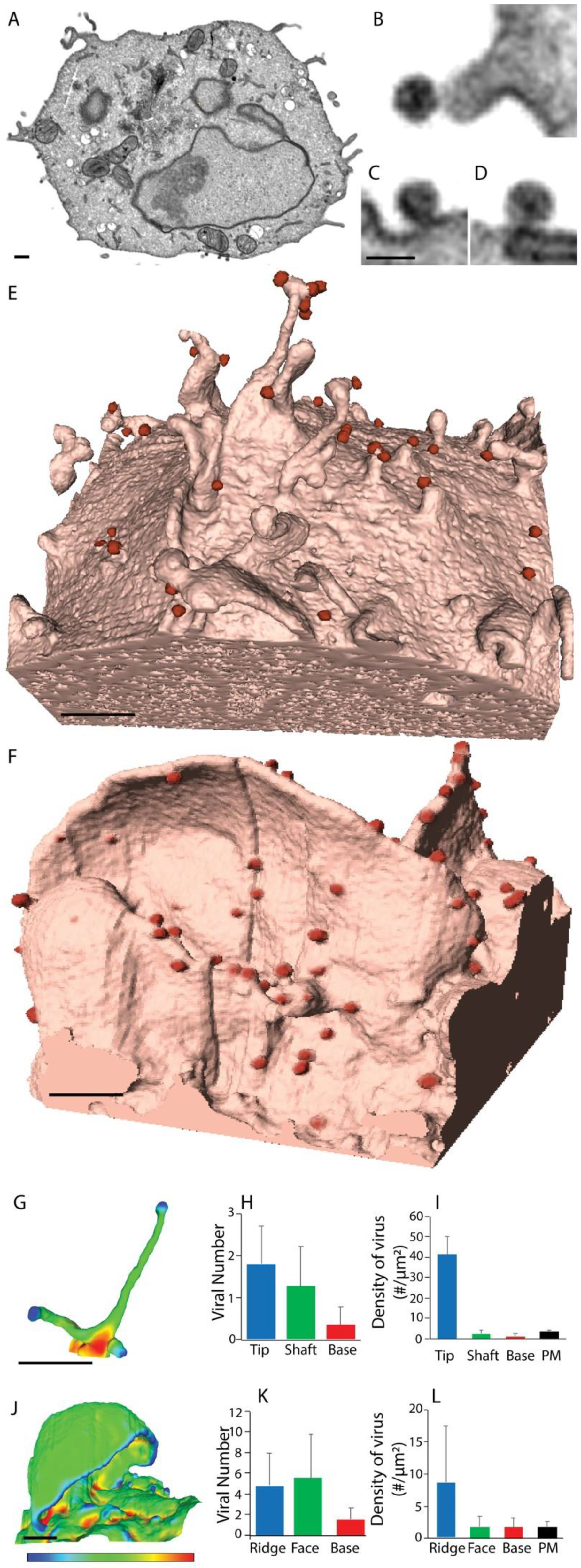
2.2. The Influence of Shifting F-Actin Structures on the Location of HIV Budding
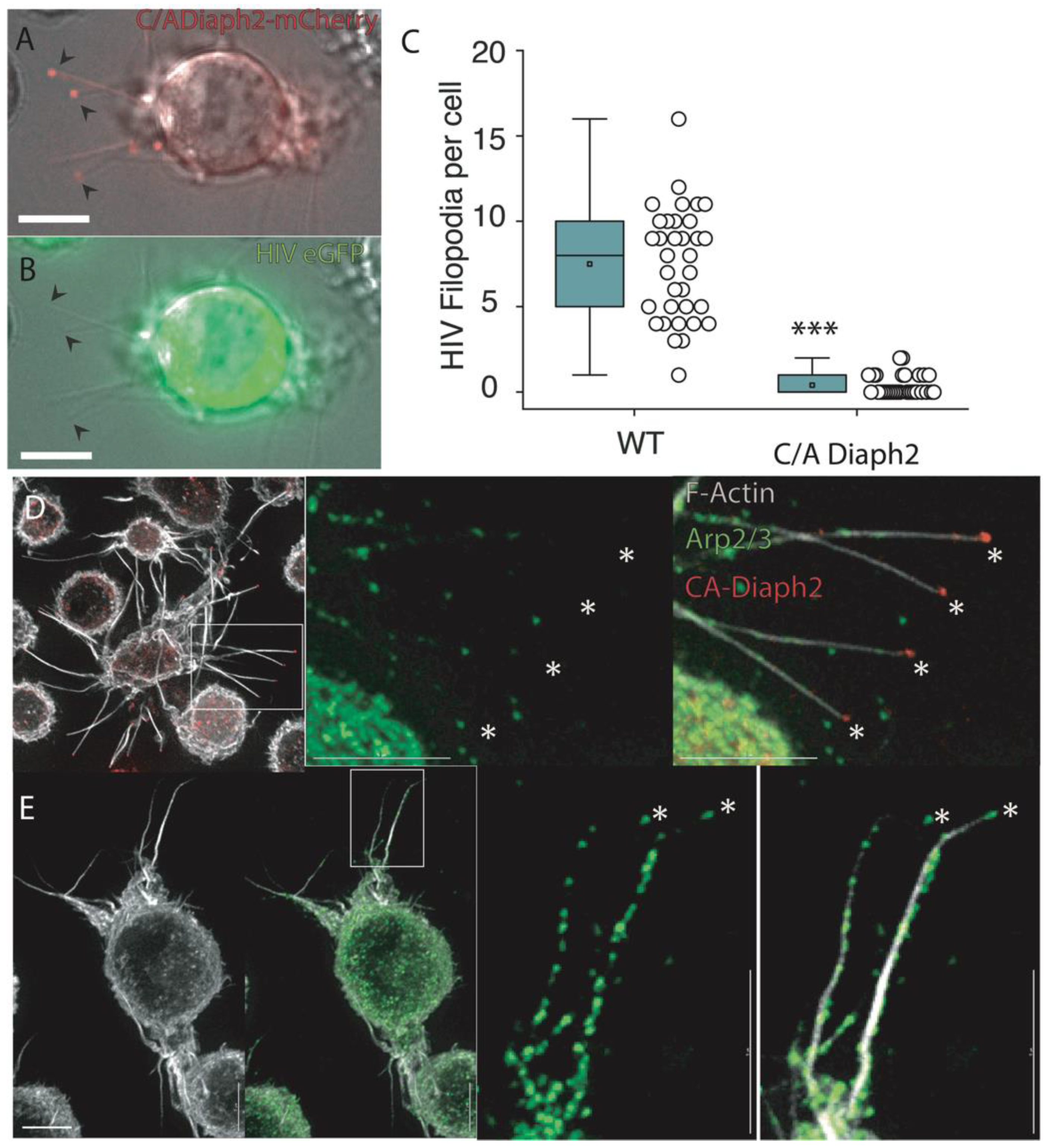
2.3. Filopodia Dominated by the Formin Diaph2 Present Positive Curvature at the Plasma Membrane but Exclude Assembling HIV Particles
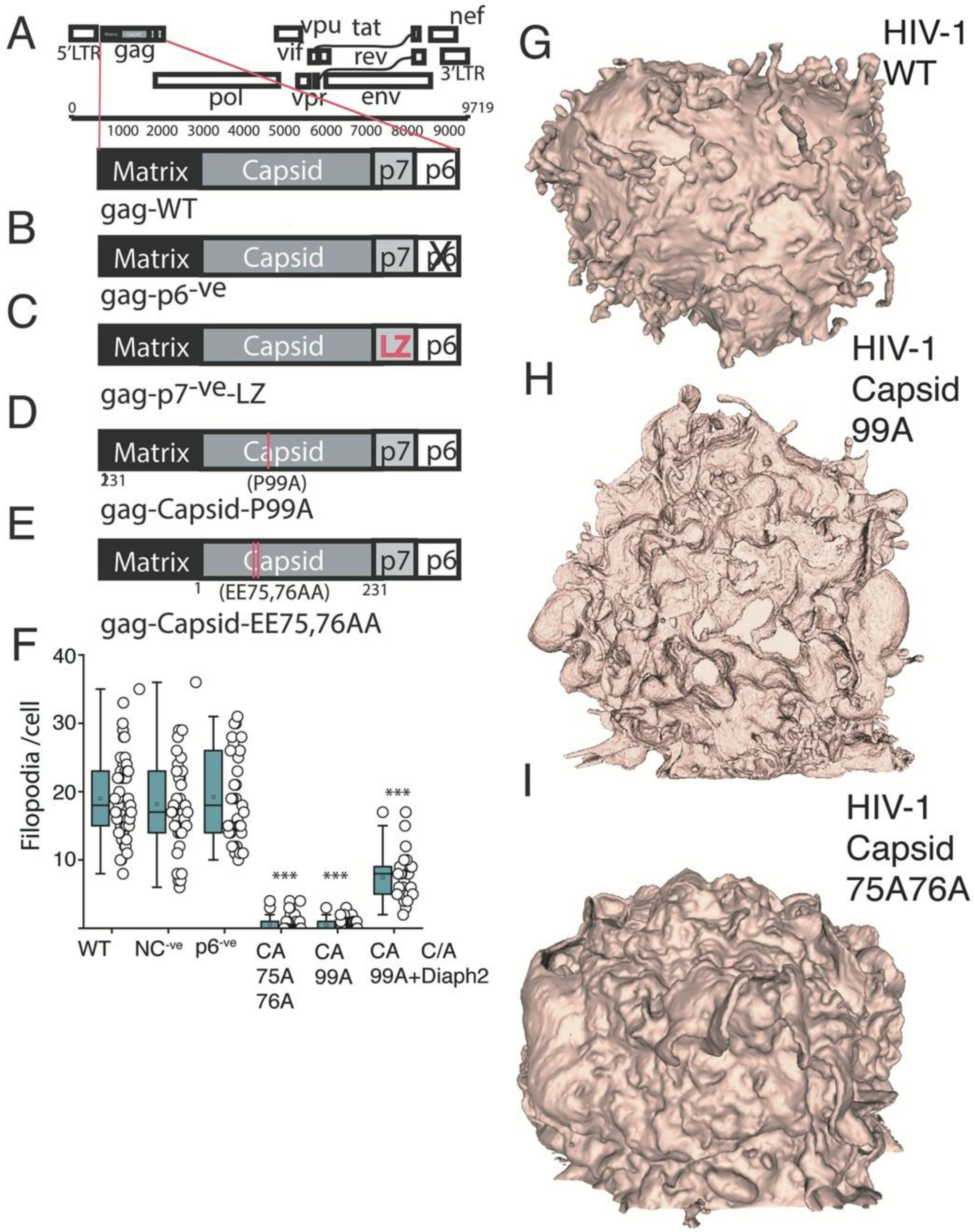
2.4. HIV Gag Can Influence Arp2/3 Dependent F-Actin Pathways
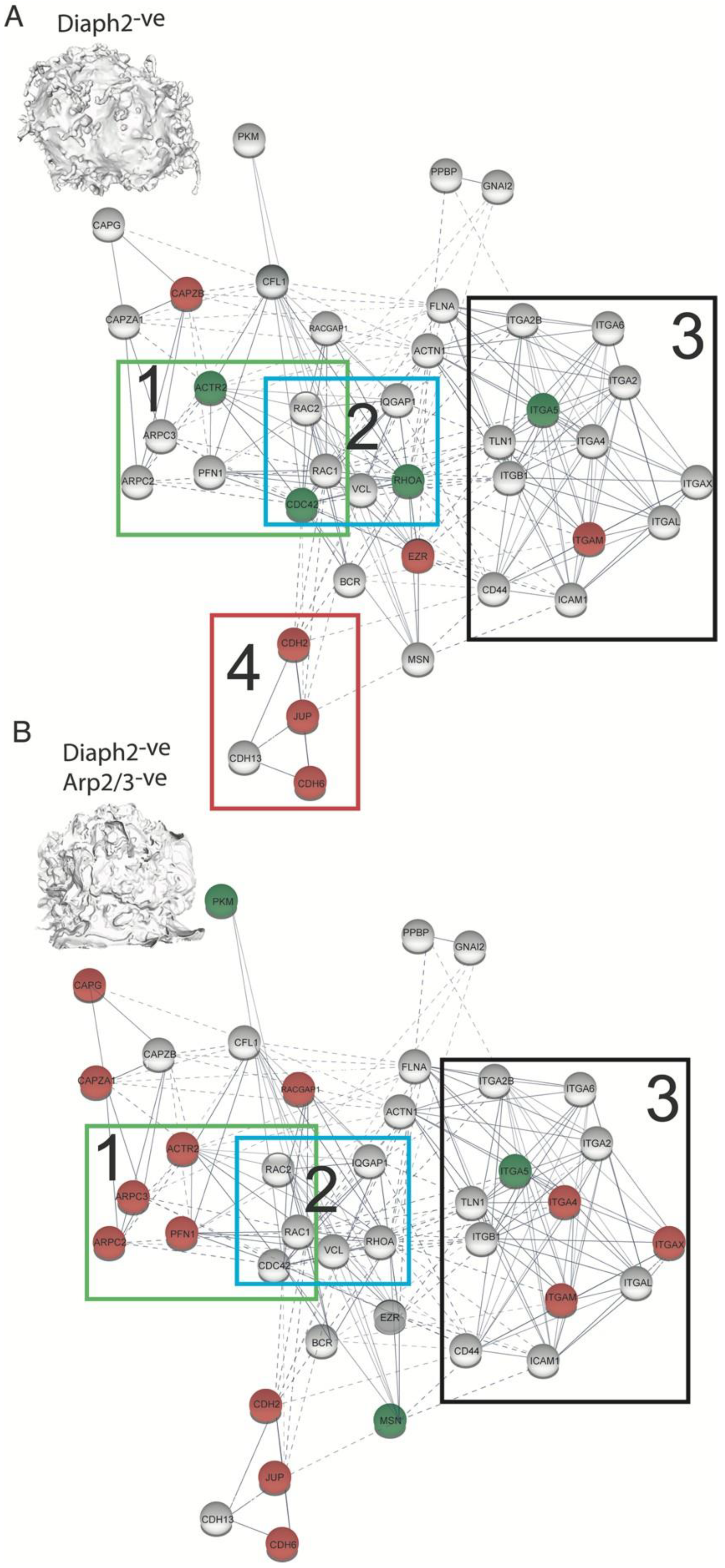
2.5. The HIV Proteome Reveals a GTPase Node Associated with Arp2/3 F-Actin Regulation
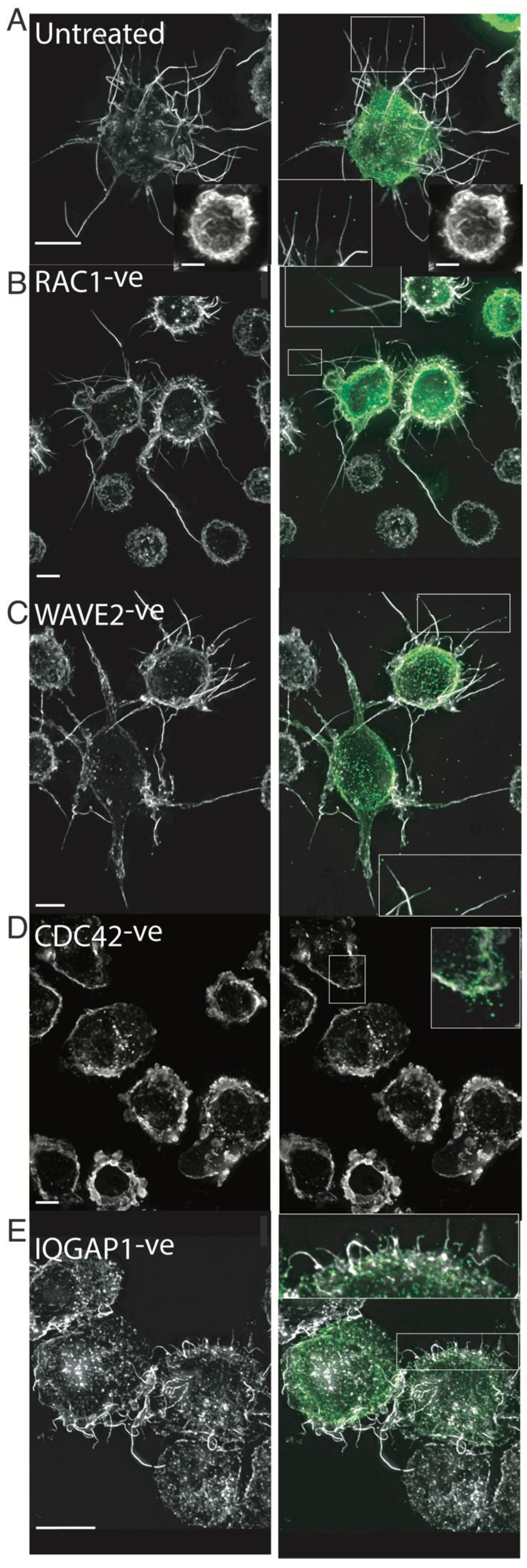
2.6. HIV Exploits the Cdc42-Arp2/3 Filopodial Pathway to Position Virus at Cell–Cell Contacts
2.7. HIV Cell–Cell Transfer Is Dependent on an Intact Cdc42-IQGAP1-Arp2/3 Pathway
3. Discussion
4. Materials and Methods
4.1. HIV Plasmid Constructs
4.2. Virus Production and Infections
4.3. Lentiviral Constructs and Production:
4.3.1. Diaph2C/A Mutant Construct
4.3.2. Lentiviral shRNA Vectors
4.3.3. Lentiviral CRISPR Vectors
4.4. Cell Culture, Genetic Modification and Infection:
4.4.1. shRNA Depletion
4.4.2. CRISPR Gene Knock Outs
4.4.3. Clonal Sorting, Depletion and Knock out Confirmation
4.4.4. Quantitative Analysis of Live Cells during Viral Transfer:
- Cell–cell contacts
- Cellular circularity vs. Gag-polarisation
- Immunofluorescence microscopy:
- Correlative FIB-SEM:
- Mass spectrometry of purified virions:
- Mass spectrometry
- Sequence database searches and protein quantification
- Statistical tests
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Robinson, R.C.; Turbedsky, K.; Kaiser, D.A.; Marchand, J.B.; Higgs, H.N.; Choe, S.; Pollard, T.D. Crystal structure of Arp2/3 complex. Science 2001, 294, 1679–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobes, C.D.; Hall, A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 1995, 81, 53–62. [Google Scholar] [CrossRef] [Green Version]
- Phillips, D.M.; Bourinbaiar, A.S. Mechanism of HIV spread from lymphocytes to epithelia. Virology 1992, 186, 261–273. [Google Scholar] [CrossRef]
- Jolly, C.; Kashefi, K.; Hollinshead, M.; Sattentau, Q.J. HIV-1 Cell to Cell Transfer across an Env-induced, Actin-dependent Synapse. J. Exp. Med. 2004, 199, 283–293. [Google Scholar] [CrossRef]
- Sundquist, W.I.; Krausslich, H.G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924. [Google Scholar] [CrossRef] [PubMed]
- Garrus, J.E.; von Schwedler, U.K.; Pornillos, O.W.; Morham, S.G.; Zavitz, K.H.; Wang, H.E.; Wettstein, D.A.; Stray, K.M.; Côté, M.; Rich, R.L.; et al. Tsg101 and the Vacuolar Protein Sorting Pathway Are Essential for HIV-1 Budding. Cell 2001, 107, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Gladnikoff, M.; Shimoni, E.; Gov, N.S.; Rousso, I. Retroviral Assembly and Budding Occur through an Actin-Driven Mechanism. Biophys. J. 2009, 97, 2419–2428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, L.-A.; De Marco, A.; Oberwinkler, H.; Habermann, A.; Briggs, J.; Kräusslich, H.-G.; Grünewald, K. Cryo Electron Tomography of Native HIV-1 Budding Sites. PLoS Pathog. 2010, 6, e1001173. [Google Scholar] [CrossRef]
- Aggarwal, A.; Iemma, T.L.; Shih, I.; Newsome, T.; McAllery, S.; Cunningham, A.; Turville, S.G. Mobilization of HIV Spread by Diaphanous 2 Dependent Filopodia in Infected Dendritic Cells. PLoS Pathog. 2012, 8, e1002762. [Google Scholar] [CrossRef] [Green Version]
- Ladinsky, M.S.; Kieffer, C.; Olson, G.; Deruaz, M.; Vrbanac, V.; Tager, A.M.; Kwon, D.S.; Bjorkman, P.J. Electron Tomography of HIV-1 Infection in Gut-Associated Lymphoid Tissue. PLoS Pathog. 2014, 10, e1003899. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Qu, Y.; Liu, Y.; Jambusaria, R.; Han, Z.; Ruthel, G.; Freedman, B.D.; Harty, R.N. Host IQGAP1 and Ebola Virus VP40 Interactions Facilitate Virus-Like Particle Egress. J. Virol. 2013, 87, 7777–7780. [Google Scholar] [CrossRef] [Green Version]
- Mattila, P.K.; Lappalainen, P. Filopodia: Molecular architecture and cellular functions. Nat. Rev. Mol. Cell Biol. 2008, 9, 446–454. [Google Scholar] [CrossRef]
- Wu, C.; Asokan, S.B.; Berginski, M.E.; Haynes, E.M.; Sharpless, N.E.; Griffith, J.D.; Gomez, S.M.; Bear, J.E. Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell 2012, 148, 973–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suraneni, P.; Rubinstein, B.; Unruh, J.R.; Durnin, M.; Hanein, D.; Li, R. The Arp2/3 complex is required for lamellipodia extension and directional fibroblast cell migration. J. Cell Biol. 2012, 197, 239–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, L.E.; Heimsath, E.G.; Higgs, H.N. Cell type-dependent mechanisms for formin-mediated assembly of filopodia. Mol. Biol. Cell 2015, 26, 4646–4659. [Google Scholar] [CrossRef]
- Eugenin, E.; Gaskill, P.; Berman, J. Tunneling nanotubes (TNT) are induced by HIV-infection of macrophages: A potential mechanism for intercellular HIV trafficking. Cell. Immunol. 2009, 254, 142–148. [Google Scholar] [CrossRef] [Green Version]
- Bourinbaiar, A.S.; Phillips, D.M. Transmission of human immunodeficiency virus from monocytes to epithelia. JAIDS J. Acquir. Immune Defic. Syndr. 1991, 4, 56–63. [Google Scholar] [CrossRef]
- Pearce-Pratt, R.; Malamud, D.; Phillips, D.M. Role of the cytoskeleton in cell-to-cell transmission of human immunodeficiency virus. J. Virol. 1994, 68, 2898–2905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higgs, H.N.; Peterson, K.J. Phylogenetic Analysis of the Formin Homology 2 Domain. Mol. Biol. Cell 2005, 16, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Narayan, K.; Danielson, C.M.; Lagarec, K.; Lowekamp, B.C.; Coffman, P.; Laquerre, A.; Phaneuf, M.W.; Hope, T.J.; Subramaniam, S. Multi-resolution correlative focused ion beam scanning electron microscopy: Applications to cell biology. J. Struct. Biol. 2014, 185, 278–284. [Google Scholar] [CrossRef] [Green Version]
- Narayan, K.; Subramaniam, S. Focused ion beams in biology. Nat. Methods 2015, 12, 1021–1031. [Google Scholar] [CrossRef]
- Felts, R.L.; Narayan, K.; Estes, J.D.; Shi, D.; Trubey, C.M.; Fu, J.; Hartnell, L.M.; Ruthel, G.T.; Schneider, D.K.; Nagashima, K.; et al. 3D visualization of HIV transfer at the virological synapse between dendritic cells and T cells. Proc. Natl. Acad. Sci. USA 2010, 107, 13336–13341. [Google Scholar] [CrossRef] [Green Version]
- Alberts, A.S. Identification of a Carboxyl-terminal Diaphanous-related Formin Homology Protein Autoregulatory Domain. J. Biol. Chem. 2001, 276, 2824–2830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lammers, M.; Rose, R.; Scrima, A.; Wittinghofer, A. The regulation of mDia1 by autoinhibition and its release by Rho GTP. EMBO J. 2005, 24, 4176–4187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Block, J.; Breitsprecher, D.; Kuhn, S.; Winterhoff, M.; Kage, F.; Geffers, R.; Duwe, P.; Rohn, J.L.; Baum, B.; Brakebusch, C.; et al. FMNL2 drives actin-based protrusion and migration downstream of Cdc42. Curr. Biol. CB 2012, 22, 1005–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Dai, R.; Tian, C.-J.; Dawson, L.; Gorelick, R.; Yu, X.-F. Interaction of the Human Immunodeficiency Virus Type 1 Nucleocapsid with Actin. J. Virol. 1999, 73, 2901–2908. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Datta, S.A.; Kim, S.H.; To, S.; Chaturvedi, S.K.; Rein, A.; Schuck, P. Nucleic acid–induced dimerization of HIV-1 Gag protein. J. Biol. Chem. 2019, 294, 16480–16493. [Google Scholar] [CrossRef] [Green Version]
- Crist, R.; Datta, S.; Stephen, A.G.; Soheilian, F.; Mirro, J.; Fisher, R.J.; Nagashima, K.; Rein, A. Assembly Properties of Human Immunodeficiency Virus Type 1 Gag-Leucine Zipper Chimeras: Implications for Retrovirus Assembly. J. Virol. 2009, 83, 2216–2225. [Google Scholar] [CrossRef] [Green Version]
- Von Schwedler, U.K.; Stray, K.M.; Garrus, J.E.; Sundquist, W.I. Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J. Virol. 2003, 77, 5439–5450. [Google Scholar] [CrossRef] [Green Version]
- Hogue, I.B.; Grover, J.R.; Soheilian, F.; Nagashima, K.; Ono, A. Gag induces the coalescence of clustered lipid rafts and tetraspanin-enriched microdomains at HIV-1 assembly sites on the plasma membrane. J. Virol. 2011, 85, 9749–9766. [Google Scholar] [CrossRef] [Green Version]
- Ott, D.E.; Coren, L.V.; Johnson, D.G.; Kane, B.P.; Sowder, R.C.; Kim, Y.D.; Fisher, R.J.; Zhou, X.Z.; Lu, K.P.; Henderson, L.E. Actin-Binding Cellular Proteins inside Human Immunodeficiency Virus Type 1. Virology 2000, 266, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Chertova, E.; Chertov, O.; Coren, L.V.; Roser, J.D.; Trubey, C.M.; Bess, J.W., Jr.; Sowder, R.C., 2nd; Barsov, E.; Hood, B.L.; Fisher, R.J.; et al. Proteomic and biochemical analysis of purified human immunodeficiency virus type 1 produced from infected monocyte-derived macrophages. J. Virol. 2006, 80, 9039–9052. [Google Scholar] [CrossRef] [Green Version]
- Stauffer, S.; Rahman, S.; De Marco, A.; Carlson, L.-A.; Glass, B.; Oberwinkler, H.; Herold, N.; Briggs, J.; Müller, B.; Grünewald, K.; et al. The Nucleocapsid Domain of Gag Is Dispensable for Actin Incorporation into HIV-1 and for Association of Viral Budding Sites with Cortical F-Actin. J. Virol. 2014, 88, 7893–7903. [Google Scholar] [CrossRef] [Green Version]
- Nelson, W.J.; Drees, F.; Yamada, S. Interaction of cadherin with the actin cytoskeleton. Novartis Found. Symp. 2005, 269, 159–168; discussion 168–177, 223–230. [Google Scholar]
- Fukata, M.; Nakagawa, M.; Itoh, N.; Kawajiri, A.; Yamaga, M.; Kuroda, S.; Kaibuchi, K. Involvement of IQGAP1, an Effector of Rac1 and Cdc42 GTPases, in Cell-Cell Dissociation during Cell Scattering. Mol. Cell. Biol. 2001, 21, 2165–2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swart-Mataraza, J.M.; Li, Z.; Sacks, D. IQGAP1 Is a Component of Cdc42 Signaling to the Cytoskeleton. J. Biol. Chem. 2002, 277, 24753–24763. [Google Scholar] [CrossRef] [Green Version]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Martinez-Quiles, N.; Eden, S.; Shibata, T.; Takeshima, F.; Shinkura, R.; Fujiwara, Y.; Bronson, R.; Snapper, S.B.; Kirschner, M.W.; et al. WAVE2 deficiency reveals distinct roles in embryogenesis and Rac-mediated actin-based motility. EMBO J. 2003, 22, 3602–3612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daste, F.; Walrant, A.; Holst, M.R.; Gadsby, J.R.; Mason, J.; Lee, J.-E.; Brook, D.; Mettlen, M.; Larsson, E.; Lee, S.F.; et al. Control of actin polymerization via the coincidence of phosphoinositides and high membrane curvature. J. Cell Biol. 2017, 216, 3745–3765. [Google Scholar] [CrossRef]
- Gallop, J.L.; Walrant, A.; Cantley, L.; Kirschner, M.W. Phosphoinositides and membrane curvature switch the mode of actin polymerization via selective recruitment of toca-1 and Snx9. Proc. Natl. Acad. Sci. USA 2013, 110, 7193–7198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabo, Y.; Santos, K.D.L.; Goff, S.P. IQGAP1 Negatively Regulates HIV-1 Gag Trafficking and Virion Production. Cell Rep. 2020, 30, 4065–4081.e4. [Google Scholar] [CrossRef]
- Brandt, D.T.; Grosse, R. Get to grips: Steering local actin dynamics with IQGAPs. EMBO Rep. 2007, 8, 1019–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolic, D.S.; Lehmann, M.; Felts, R.; Garcia, E.; Blanchet, F.P.; Subramaniam, S.; Piguet, V. HIV-1 activates Cdc42 and induces membrane extensions in immature dendritic cells to facilitate cell-to-cell virus propagation. Blood 2011, 118, 4841–4852. [Google Scholar] [CrossRef]
- Fukata, M.; Kuroda, S.; Fujii, K.; Nakamura, T.; Shoji, I.; Matsuura, Y.; Okawa, K.; Iwamatsu, A.; Kikuchi, A.; Kaibuchi, K. Regulation of Cross-linking of Actin Filament by IQGAP1, a Target for Cdc42. J. Biol. Chem. 1997, 272, 29579–29583. [Google Scholar] [CrossRef] [Green Version]
- Bendjennat, M.; Saffarian, S. The Race against Protease Activation Defines the Role of ESCRTs in HIV Budding. PLoS Pathog. 2016, 12, e1005657. [Google Scholar] [CrossRef] [PubMed]
- Damsky, C.H.; Sheffield, J.B.; Tuszynski, G.P.; Warren, L. Is there a role for actin in virus budding? J. Cell Biol. 1977, 75, 593–605. [Google Scholar] [CrossRef]
- Jouvenet, N.; Windsor, M.; Rietdorf, J.; Hawes, P.; Monaghan, P.; Way, M.; Wileman, T. African swine fever virus induces filopodia-like projections at the plasma membrane. Cell. Microbiol. 2006, 8, 1803–1811. [Google Scholar] [CrossRef] [PubMed]
- Kolesnikova, L.; Bohil, A.B.; Cheney, R.E.; Becker, S. Budding of Marburgvirus is associated with filopodia. Cell. Microbiol. 2007, 9, 939–951. [Google Scholar] [CrossRef]
- Kolesnikova, L.; Heck, S.; Matrosovich, T.; Klenk, H.D.; Becker, S.; Matrosovich, M. Influenza virus budding from the tips of cellular microvilli in differentiated human airway epithelial cells. J. Gen. Virol. 2013, 94, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, J.E.; Hutchinson, J.A.; Jackson, W.; Grose, C. Egress of light particles among filopodia on the surface of Varicella-Zoster virus-infected cells. J. Virol. 2008, 82, 2821–2835. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.; Baginski, J.; Hassan, S.F.; Volin, M.; Shukla, D.; Tiwari, V. Filopodia and Viruses: An Analysis of Membrane Processes in Entry Mechanisms. Front. Microbiol. 2016, 7, 300. [Google Scholar]
- Lemichez, E.; Aktories, K. Hijacking of Rho GTPases during bacterial infection. Exp. Cell Res. 2013, 319, 2329–2336. [Google Scholar] [CrossRef]
- Van den Broeke, C.; Jacob, T.; Favoreel, H.W. Rho’ing in and out of cells: Viral interactions with Rho GTPase signaling. Small GTPases 2014, 5, e28318. [Google Scholar] [CrossRef] [PubMed]
- Gouin, E.; Welch, M.D.; Cossart, P. Actin-based motility of intracellular pathogens. Curr. Opin. Microbiol. 2005, 8, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Small, J.V. Pushing with actin: From cells to pathogens. Biochem. Soc. Trans. 2015, 43, 84–91. [Google Scholar] [CrossRef]
- Welch, M.D.; Way, M. Arp2/3-mediated actin-based motility: A tail of pathogen abuse. Cell Host Microbe 2013, 14, 242–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; White, C.D.; Sacks, D.B. IQGAP1 in microbial pathogenesis: Targeting the actin cytoskeleton. FEBS Lett. 2011, 585, 723–729. [Google Scholar] [CrossRef] [Green Version]
- Leung, J.; Yueh, A.; Appah, F.S.K.; Yuan, B.; Santos, K.D.L.; Goff, S.P. Interaction of Moloney murine leukemia virus matrix protein with IQGAP. EMBO J. 2006, 25, 2155–2166. [Google Scholar] [CrossRef] [Green Version]
- Gladue, D.; Holinka, L.; Fernandez-Sainz, I.; Prarat, M.; O’Donnell, V.; Vepkhvadze, N.; Lu, Z.; Risatti, G.; Borca, M. Interaction between Core protein of classical swine fever virus with cellular IQGAP1 protein appears essential for virulence in swine. Virology 2011, 412, 68–74. [Google Scholar] [CrossRef]
- Dolnik, O.; Kolesnikova, L.; Welsch, S.; Strecker, T.; Schudt, G.; Becker, S. Interaction with Tsg101 Is Necessary for the Efficient Transport and Release of Nucleocapsids in Marburg Virus-Infected Cells. PLoS Pathog. 2014, 10, e1004463. [Google Scholar] [CrossRef] [Green Version]
- Dolnik, O.; Stevermann, L.; Kolesnikova, L.; Becker, S. Marburg virus inclusions: A virus-induced microcompartment and interface to multivesicular bodies and the late endosomal compartment. Eur. J. Cell Biol. 2015, 94, 323–331. [Google Scholar] [CrossRef]
- Mehedi, M.; Collins, P.L.; Buchholz, U.J. A novel host factor for human respiratory syncytial virus. Commun. Integr. Biol. 2017, 10, e1319025. [Google Scholar] [CrossRef] [Green Version]
- Mehedi, M.; McCarty, T.; Martin, S.E.; Le Nouën, C.; Buehler, E.; Chen, Y.-C.; Smelkinson, M.; Ganesan, S.; Fischer, E.R.; Brock, L.G.; et al. Actin-Related Protein 2 (ARP2) and Virus-Induced Filopodia Facilitate Human Respiratory Syncytial Virus Spread. PLoS Pathog. 2016, 12, e1006062. [Google Scholar] [CrossRef]
- Martinez, M.G.; Kielian, M. Intercellular Extensions Are Induced by the Alphavirus Structural Proteins and Mediate Virus Transmission. PLoS Pathog. 2016, 12, e1006061. [Google Scholar] [CrossRef] [Green Version]
- Sherer, N.M.; Lehmann, M.J.; Soto, L.F.J.; Horensavitz, C.; Pypaert, M.; Mothes, W. Retroviruses can establish filopodial bridges for efficient cell-to-cell transmission. Nat. Cell Biol. 2007, 9, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cifuentes-Muñoz, N.; Dutch, R.E.; Cattaneo, R. Direct cell-to-cell transmission of respiratory viruses: The fast lanes. PLoS Pathog. 2018, 14, e1007015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziv, N.; Smith, S.J. Evidence for a Role of Dendritic Filopodia in Synaptogenesis and Spine Formation. Neuron 1996, 17, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Niell, C.M.; Meyer, M.; Smith, S.J. In vivo imaging of synapse formation on a growing dendritic arbor. Nat. Neurosci. 2004, 7, 254–260. [Google Scholar] [CrossRef]
- Vasioukhin, V.; Bauer, C.; Yin, M.; Fuchs, E. Directed Actin Polymerization Is the Driving Force for Epithelial Cell–Cell Adhesion. Cell 2000, 100, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Menna, E.; Fossati, G.; Scita, G.; Matteoli, M. From filopodia to synapses: The role of actin-capping and anti-capping proteins. Eur. J. Neurosci. 2011, 34, 1655–1662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mothes, W.; Sherer, N.M.; Jin, J.; Zhong, P. Virus Cell-to-Cell Transmission. J. Virol. 2010, 84, 8360–8368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demirov, D.G.; Ono, A.; Orenstein, J.M.; Freed, E.O. Overexpression of the N-terminal domain of TSG101 inhibits HIV-1 budding by blocking late domain function. Proc. Natl. Acad. Sci. USA 2002, 99, 955–960. [Google Scholar] [CrossRef] [Green Version]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Concordet, J.-P.; Haeussler, M. CRISPOR: Intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018, 46, W242–W245. [Google Scholar] [CrossRef]
- Guschin, D.Y.; Waite, A.J.; Katibah, G.E.; Miller, J.C.; Holmes, M.C.; Rebar, E.J. A Rapid and General Assay for Monitoring Endogenous Gene Modification. Methods Mol. Bio. 2010, 649, 247–256. [Google Scholar]
- Meijering, E.; Dzyubachyk, O.; Smal, I. Methods for Cell and Particle Tracking. Methods Enzymol. 2012, 504, 183–200. [Google Scholar] [PubMed]
- Kremer, J.R.; Mastronarde, D.N.; McIntosh, J.R. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 1996, 116, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Fedorov, A.; Beichel, R.; Kalpathy-Cramer, J.; Finet, J.; Fillion-Robin, J.-C.; Pujol, S.; Bauer, C.; Jennings, D.; Fennessy, F.; Sonka, M.; et al. 3D Slicer as an image computing platform for the Quantitative Imaging Network. Magn. Reson. Imaging 2012, 30, 1323–1341. [Google Scholar] [CrossRef] [Green Version]
- Hevchenko, A.; Jensen, O.N.; Podtelejnikov, A.V.; Sagliocco, F.; Wilm, M.; Vorm, O.; Mortensen, P.; Shevchenko, A.; Boucherie, H.; Mann, M. Linking genome and proteome by mass spectrometry: Large-scale identification of yeast proteins from two dimensional gels. Proc. Natl. Acad. Sci. USA 1996, 93, 14440–14445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart-Smith, G.; Raftery, M.J. Detection and characterization of low abundance glycopeptides via higher-energy C-trap dissociation and orbitrap mass analysis. J. Am. Soc. Mass. Spectrom. 2012, 23, 124–140. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aggarwal, A.; Stella, A.O.; Henry, C.C.; Narayan, K.; Turville, S.G. Embedding of HIV Egress within Cortical F-Actin. Pathogens 2022, 11, 56. https://doi.org/10.3390/pathogens11010056
Aggarwal A, Stella AO, Henry CC, Narayan K, Turville SG. Embedding of HIV Egress within Cortical F-Actin. Pathogens. 2022; 11(1):56. https://doi.org/10.3390/pathogens11010056
Chicago/Turabian StyleAggarwal, Anupriya, Alberto Ospina Stella, Catherine C. Henry, Kedar Narayan, and Stuart G. Turville. 2022. "Embedding of HIV Egress within Cortical F-Actin" Pathogens 11, no. 1: 56. https://doi.org/10.3390/pathogens11010056