
De-Coding the Contributions of the Viral RNAs to Alphaviral Pathogenesis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Background
2. The Contribution of Alphaviral RNAs to Pathogenesis, an Emerging Frontier
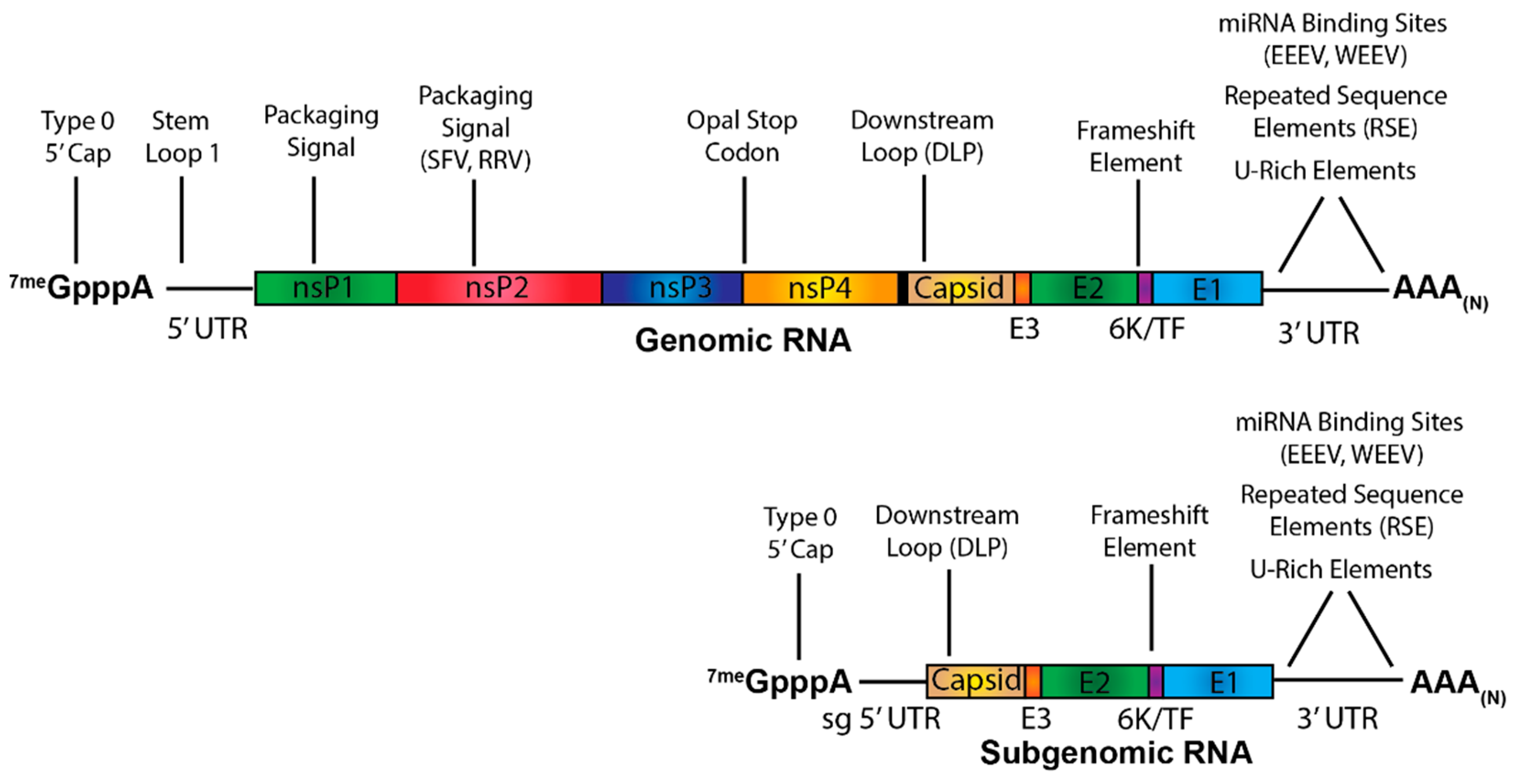
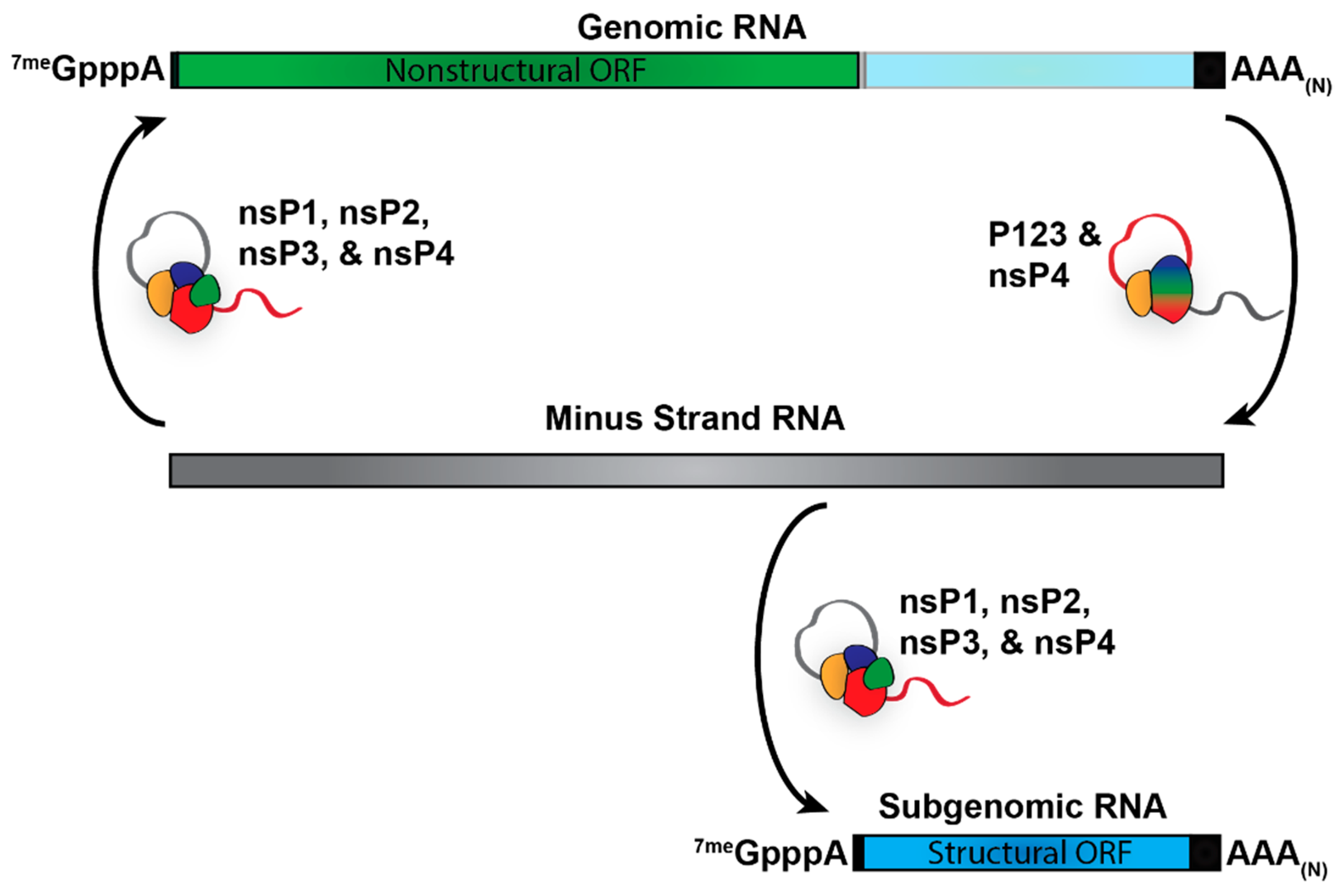
3. Alphaviral RNA Species and Viral Replication/Transcription
4. Contributions of the Alphaviral Positive-Sense RNAs to Pathogenesis
4.1. The Contribution of Non-Templated Features of Alphaviral RNAs
4.2. The Contribution of Untranslated Regions to Alphaviral Pathogenesis
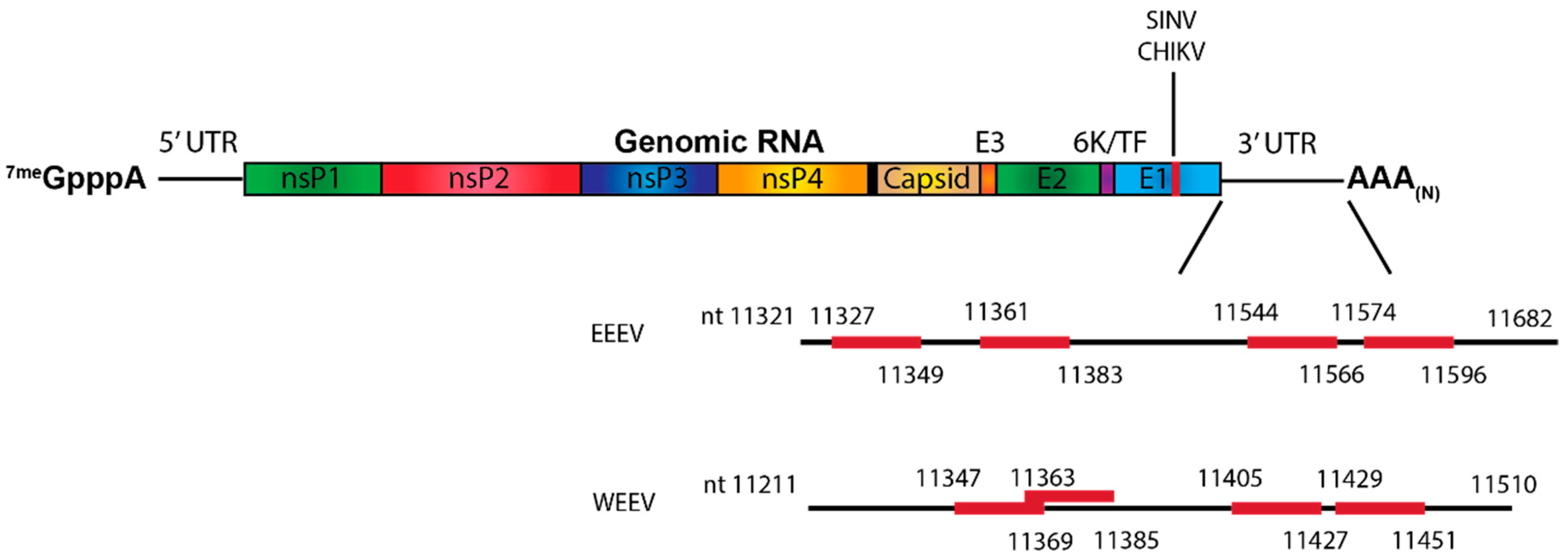
4.2.1. The 5′ Untranslated Regions and Adjacent Sequences
4.2.2. The 3′ Untranslated Region
4.3. Virulence Traits of the Nonstructural and Structural Open Reading Frames
4.3.1. Dinucleotide Motif Biases in Alphaviral Pathogenesis
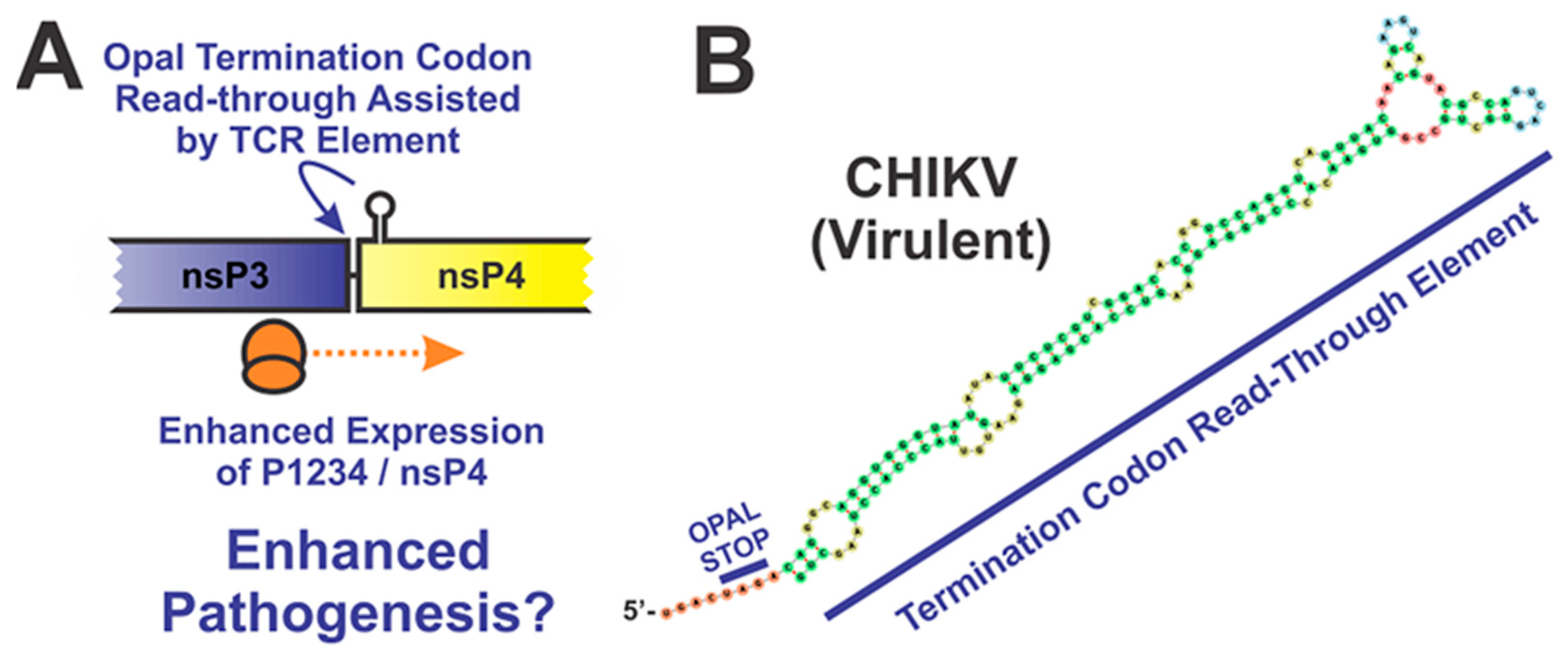
4.3.2. The Opal Stop Codon
4.3.3. Secondary Structures of the Alphaviruses Open Reading Frames
4.4. Protein:vRNA Interactions
5. Contributions of Other Alphaviral RNAs to Pathogenesis
5.1. The Contribution of the Minus-Strand RNA to Alphaviral Pathogenesis
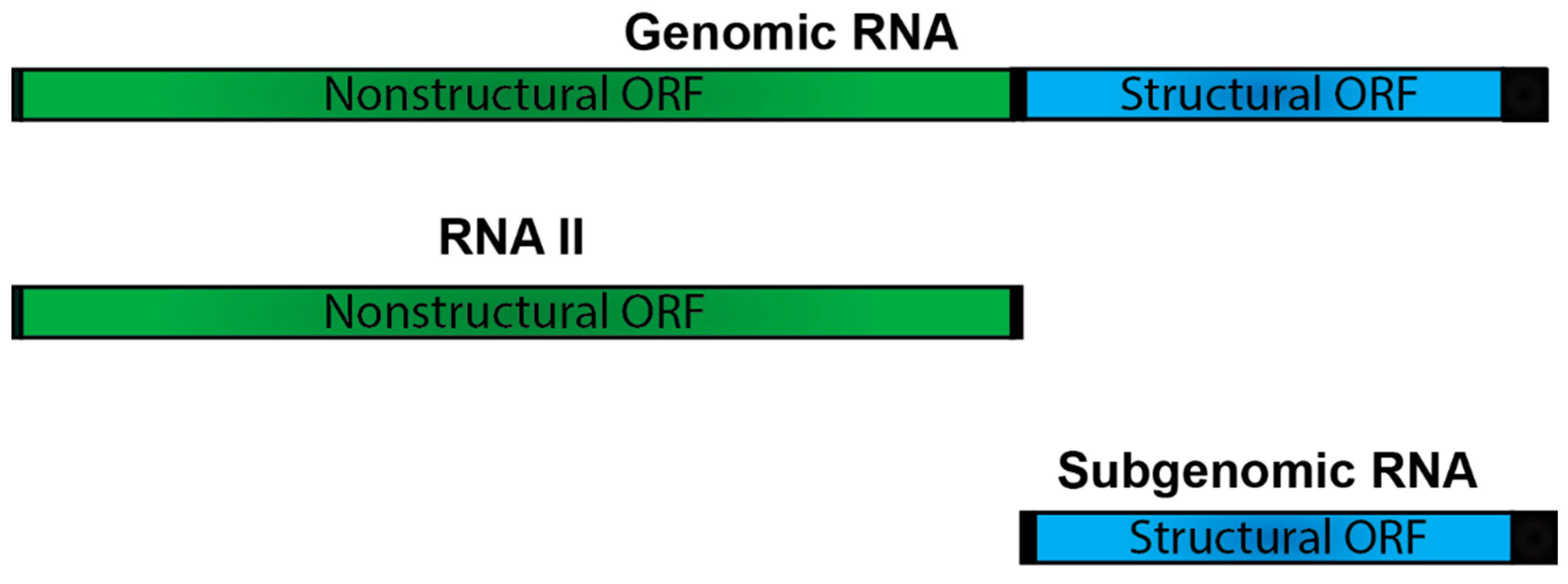
5.2. The Alphaviral RNA II- a Consistent Curiosity
5.3. Defective Viral RNA Species
6. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kurkela, S.; Helve, T.; Vaheri, A.; Vapalahti, O. Arthritis and arthralgia three years after Sindbis virus infection: Clinical follow-up of a cohort of 49 patients. Scand. J. Infect. Dis. 2008, 40, 167–173. [Google Scholar] [CrossRef]
- Kurkela, S.; Manni, T.; Myllynen, J.; Vaheri, A.; Vapalahti, O. Clinical and Laboratory Manifestations of Sindbis Virus Infection: Prospective Study, Finland, 2002–2003. J. Infect. Dis. 2005, 191, 1820–1829. [Google Scholar] [CrossRef]
- Kurkela, S.; Rätti, O.; Huhtamo, E.; Uzcátegui, N.Y.; Nuorti, J.P.; Laakkonen, J.; Manni, T.; Helle, P.; Vaheri, A.; Vapalahti, O. Sindbis Virus Infection in Resident Birds, Migratory Birds, and Humans, Finland. Emerg. Infect. Dis. 2008, 14, 41–47. [Google Scholar] [CrossRef]
- Sissoko, D.; Malvy, D.; Ezzedine, K.; Renault, P.; Moscetti, F.; Ledrans, M.; Pierre, V. Post-Epidemic Chikungunya Disease on Reunion Island: Course of Rheumatic Manifestations and Associated Factors over a 15-Month Period. PLoS Negl. Trop. Dis. 2009, 3, e389. [Google Scholar] [CrossRef] [Green Version]
- Farnon, E.C.; Sejvar, J.J.; Staples, J.E. Severe disease manifestations associated with acute chikungunya virus infection*. Crit. Care Med. 2008, 36, 2682–2683. [Google Scholar] [CrossRef]
- Cardona-Ospina, J.A.; Villamil-Gómez, W.E.; Jimenez-Canizales, C.E.; Castañeda, D.M.; Rodríguez-Morales, A.J. Estimating the burden of disease and the economic cost attributable to chikungunya, Colombia, 2014. Trans. R. Soc. Trop. Med. Hyg. 2015, 109, 793–802. [Google Scholar] [CrossRef]
- Seyler, T.; Hutin, Y.; Ramanchandran, V.; Ramakrishnan, R.; Manickam, P.; Murhekar, M. Estimating the burden of disease and the economic cost attributable to chikungunya, Andhra Pradesh, India, 2005–2006. Trans. R. Soc. Trop. Med. Hyg. 2010, 104, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Calisher, C.H. Medically important arboviruses of the United States and Canada. Clin. Microbiol. Rev. 1994, 7, 89–116. [Google Scholar] [CrossRef]
- De La Monte, S.M.; Bonilla, N.J.; De Urdaneta, A.G.; Hutchins, G.M.; Castro, F. The Systemic Pathology of Venezuelan Equine Encephalitis Virus Infection in Humans. Am. J. Trop. Med. Hyg. 1985, 34, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Ronca, S.E.; Dineley, K.T.; Paessler, S. Neurological Sequelae Resulting from Encephalitic Alphavirus Infection. Front. Microbiol. 2016, 7, 959. [Google Scholar] [CrossRef] [PubMed]
- Griffin, D.E. Emergence and re-emergence of viral diseases of the central nervous system. Prog. Neurobiol. 2010, 91, 95–101. [Google Scholar] [CrossRef]
- Steele, K.R.D.; Glass, P.; Hart, M.; Ludwig, G.; Pratt, W.; Parker, M.; Smith, J. Chapter 12: Alphavirus Encephalitides. In Medical Aspects of Biological Warfare; Reed, W., Ed.; Department of the Army: Arlington, VI, USA, 2007; pp. 241–270. [Google Scholar]
- Hardy, W.R.; Strauss, J.H. Processing the nonstructural polyproteins of sindbis virus: Nonstructural proteinase is in the C-terminal half of nsP2 and functions both in cis and in trans. J. Virol. 1989, 63, 4653–4664. [Google Scholar] [CrossRef] [Green Version]
- Rupp, J.C.; Sokoloski, K.J.; Gebhart, N.N.; Hardy, R.W. Alphavirus RNA synthesis and non-structural protein functions. J. Gen. Virol. 2015, 96, 2483–2500. [Google Scholar] [CrossRef]
- Ghosh, A.; Lima, C.D. Enzymology of RNA cap synthesis. Wiley Interdiscip. Rev. RNA 2010, 1, 152–172. [Google Scholar] [CrossRef] [Green Version]
- Akhrymuk, I.; Frolov, I.; Frolova, E. Both RIG-I and MDA5 detect alphavirus replication in concentration-dependent mode. Virology 2016, 487, 230–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokoloski, K.J.; Haist, K.C.; Morrison, T.E.; Mukhopadhyay, S.; Hardy, R.W. Noncapped Alphavirus Genomic RNAs and Their Role during Infection. J. Virol. 2015, 89, 6080–6092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, X.; Linehan, M.M.; Iwasaki, A.; Pyle, A.M. RIG-I Selectively Discriminates against 5′-Monophosphate RNA. Cell Rep. 2019, 26, 2019–2027.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapointe, A.T.; Landers, V.D.; Westcott, C.E.; Sokoloski, K.J. Production of Noncapped Genomic RNAs Is Critical to Sindbis Virus Disease and Pathogenicity. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Strauss, J.H.; Strauss, E.G. The alphaviruses: Gene expression, replication, and evolution. Microbiol. Rev. 1994, 58, 491–562. [Google Scholar] [CrossRef]
- Hyde, J.L.; Chen, R.; Trobaugh, D.; Diamond, M.S.; Weaver, S.C.; Klimstra, W.B.; Wilusz, J. The 5′ and 3′ ends of alphavirus RNAs—Non-coding is not non-functional. Virus Res. 2015, 206, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Frolov, I.; Hardy, R.; Rice, C.M. Cis-acting RNA elements at the 5′ end of Sindbis virus genome RNA regulate minus- and plus-strand RNA synthesis. RNA 2001, 7, 1638–1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, L.J.; Wang, J.-G.; Davis, N.L.; Johnston, R.E. Role of Alpha/Beta Interferon in Venezuelan Equine Encephalitis Virus Pathogenesis: Effect of an Attenuating Mutation in the 5′ Untranslated Region. J. Virol. 2001, 75, 3706–3718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinney, R.M.; Chang, G.J.; Tsuchiya, K.R.; Sneider, J.M.; Roehrig, J.; Woodward, T.M.; Trent, D.W. Attenuation of Venezuelan equine encephalitis virus strain TC-83 is encoded by the 5′-noncoding region and the E2 envelope glycoprotein. J. Virol. 1993, 67, 1269–1277. [Google Scholar] [CrossRef] [Green Version]
- Spotts, D.R.; Reich, R.M.; Kalkhan, M.A.; Kinney, R.M.; Roehrig, J. Resistance to Alpha/Beta Interferons Correlates with the Epizootic and Virulence Potential of Venezuelan Equine Encephalitis Viruses and Is Determined by the 5′ Noncoding Region and Glycoproteins. J. Virol. 1998, 72, 10286–10291. [Google Scholar] [CrossRef] [Green Version]
- Logue, C.H.; Sheahan, B.J.; Atkins, G.J. The 5′ untranslated region as a pathogenicity determinant of Semliki Forest virus in mice. Virus Genes 2008, 36, 313–321. [Google Scholar] [CrossRef]
- Kobiler, D.; Rice, C.M.; Brodie, C.; Shahar, A.; Dubuisson, J.; Halevy, M.; Lustig, S. A Single Nucleotide Change in the 5′ Noncoding Region of Sindbis Virus Confers Neurovirulence in Rats. J. Virol. 1999, 73, 10440–10446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, R.J.; Griffin, D.E.; Zhang, H.; Niesters, H.G.; Strauss, J.H. Attenuation of Sindbis virus neurovirulence by using defined mutations in nontranslated regions of the genome RNA. J. Virol. 1992, 66, 7121–7127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyde, J.L.; Gardner, C.L.; Kimura, T.; White, J.P.; Liu, G.; Trobaugh, D.; Huang, C.; Tonelli, M.; Paessler, S.; Takeda, K.; et al. A Viral RNA Structural Element Alters Host Recognition of Nonself RNA. Science 2014, 343, 783–787. [Google Scholar] [CrossRef] [Green Version]
- Reynaud, J.M.; Kim, D.Y.; Atasheva, S.; Rasalouskaya, A.; White, J.P.; Diamond, M.S.; Weaver, S.C.; Frolova, E.; Frolov, I. IFIT1 Differentially Interferes with Translation and Replication of Alphavirus Genomes and Promotes Induction of Type I Interferon. PLoS Pathog. 2015, 11, e1004863. [Google Scholar] [CrossRef] [Green Version]
- Nickens, D.G.; Hardy, R.W. Structural and functional analyses of stem–loop 1 of the Sindbis virus genome. Virology 2008, 370, 158–172. [Google Scholar] [CrossRef] [Green Version]
- Frolov, I.; Schlesinger, S. Translation of Sindbis virus mRNA: Analysis of sequences downstream of the initiating AUG codon that enhance translation. J. Virol. 1996, 70, 1182–1190. [Google Scholar] [CrossRef] [Green Version]
- Ventoso, I. Adaptive Changes in Alphavirus mRNA Translation Allowed Colonization of Vertebrate Hosts. J. Virol. 2012, 86, 9484–9494. [Google Scholar] [CrossRef] [Green Version]
- Lemaire, P.A.; Anderson, E.; Lary, J.; Cole, J.L. Mechanism of PKR Activation by dsRNA. J. Mol. Biol. 2008, 381, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Toribio, R.; López, R.T.; Boskovic, J.; Ventoso, I. An RNA trapping mechanism in Alphavirus mRNA promotes ribosome stalling and translation initiation. Nucleic Acids Res. 2016, 44, 4368–4380. [Google Scholar] [CrossRef]
- Sanz, M.A.; Almela, E.G.; Carrasco, L. Translation of Sindbis Subgenomic mRNA is Independent of eIF2, eIF2A and eIF2D. Sci. Rep. 2017, 7, srep43876. [Google Scholar] [CrossRef] [Green Version]
- Ventoso, I.; Sanz, M.A.; Molina, S.; Berlanga, J.J.; Carrasco, L.; Esteban, M. Translational resistance of late alphavirus mRNA to eIF2alpha phosphorylation: A strategy to overcome the antiviral effect of protein kinase PKR. Genes Dev. 2006, 20, 87–100. [Google Scholar] [CrossRef] [Green Version]
- Toribio, R.; Díaz-López, I.; Berlanga, J.J.; Molina-Jiménez, F.; Majano, P.; Ventoso, I. Naturally Occurring and Engineered Alphaviruses Sensitive to Double-Stranded-RNA-Activated Protein Kinase Show Restricted Translation in Mammalian Cells, Increased Sensitivity to Interferon, and Marked Oncotropism. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Pfeffer, M.; Kinney, R.M.; Rügerkaadena, O. The Alphavirus 3′-Nontranslated Region: Size Heterogeneity and Arrangement of Repeated Sequence Elements. Virology 1998, 240, 100–108. [Google Scholar] [CrossRef] [Green Version]
- Hardy, R.W.; Rice, C.M. Requirements at the 3′ End of the Sindbis Virus Genome for Efficient Synthesis of Minus-Strand RNA. J. Virol. 2005, 79, 4630–4639. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Wang, E.; Tsetsarkin, K.A.; Weaver, S.C. Chikungunya Virus 3′ Untranslated Region: Adaptation to Mosquitoes and a Population Bottleneck as Major Evolutionary Forces. PLoS Pathog. 2013, 9, e1003591. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Moreno, M.; Sanz, M.A.; Carrasco, L. A Viral mRNA Motif at the 3′-Untranslated Region that Confers Translatability in a Cell-Specific Manner. Implications for Virus Evolution. Sci. Rep. 2016, 6, srep19217. [Google Scholar] [CrossRef]
- Filomatori, C.V.; Merwaiss, F.; Bardossy, E.S.; Alvarez, D.E. Impact of alphavirus 3′UTR plasticity on mosquito transmission. Semin. Cell Dev. Biol. 2021, 111, 148–155. [Google Scholar] [CrossRef]
- Morley, V.J.; Noval, M.G.; Chen, R.; Weaver, S.C.; Vignuzzi, M.; Stapleford, K.A.; Turner, P.E. Chikungunya virus evolution following a large 3′UTR deletion results in host-specific molecular changes in protein-coding regions. Virus Evol. 2018, 4, vey012. [Google Scholar] [CrossRef] [Green Version]
- Hawman, D.W.; Carpentier, K.S.; Fox, J.; May, N.A.; Sanders, W.; Montgomery, S.A.; Moorman, N.J.; Diamond, M.S.; Morrison, T.E. Mutations in the E2 Glycoprotein and the 3′ Untranslated Region Enhance Chikungunya Virus Virulence in Mice. J. Virol. 2017, 91, e00816-17. [Google Scholar] [CrossRef] [Green Version]
- Garneau, N.L.; Sokoloski, K.J.; Opyrchal, M.; Neff, C.P.; Wilusz, C.J.; Wilusz, J. The 3′ Untranslated Region of Sindbis Virus Represses Deadenylation of Viral Transcripts in Mosquito and Mammalian Cells. J. Virol. 2008, 82, 880–892. [Google Scholar] [CrossRef] [Green Version]
- Sokoloski, K.J.; Dickson, A.M.; Chaskey, E.L.; Garneau, N.L.; Wilusz, C.J.; Wilusz, J. Sindbis Virus Usurps the Cellular HuR Protein to Stabilize Its Transcripts and Promote Productive Infections in Mammalian and Mosquito Cells. Cell Host Microbe 2010, 8, 196–207. [Google Scholar] [CrossRef] [Green Version]
- Dickson, A.M.; Anderson, J.R.; Barnhart, M.D.; Sokoloski, K.J.; Oko, L.; Opyrchal, M.; Galanis, E.; Wilusz, C.J.; Morrison, T.E.; Wilusz, J. Dephosphorylation of HuR Protein during Alphavirus Infection Is Associated with HuR Relocalization to the Cytoplasm*. J. Biol. Chem. 2012, 287, 36229–36238. [Google Scholar] [CrossRef] [Green Version]
- Trobaugh, D.; Gardner, C.L.; Sun, C.; Haddow, A.D.; Wang, E.; Chapnik, E.; Mildner, A.; Weaver, S.C.; Ryman, K.D.; Klimstra, W.B. RNA viruses can hijack vertebrate microRNAs to suppress innate immunity. Nature 2014, 506, 245–248. [Google Scholar] [CrossRef] [Green Version]
- Jopling, C.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of Hepatitis C Virus RNA Abundance by a Liver-Specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [Green Version]
- Shimakami, T.; Yamane, D.; Jangra, R.K.; Kempf, B.J.; Spaniel, C.; Barton, D.J.; Lemon, S.M. Stabilization of hepatitis C virus RNA by an Ago2-miR-122 complex. Proc. Natl. Acad. Sci. USA 2012, 109, 941–946. [Google Scholar] [CrossRef] [Green Version]
- Trobaugh, D.W.; Sun, C.; Bhalla, N.; Gardner, C.L.; Dunn, M.D.; Klimstra, W.B. Cooperativity between the 3′ untranslated region microRNA binding sites is critical for the virulence of eastern equine encephalitis virus. PLoS Pathog. 2019, 15, e1007867. [Google Scholar] [CrossRef] [Green Version]
- Gardner, C.L.; Burke, C.W.; Tesfay, M.Z.; Glass, P.J.; Klimstra, W.B.; Ryman, K.D. Eastern and Venezuelan Equine Encephalitis Viruses Differ in Their Ability To Infect Dendritic Cells and Macrophages: Impact of Altered Cell Tropism on Pathogenesis. J. Virol. 2008, 82, 10634–10646. [Google Scholar] [CrossRef] [Green Version]
- López, P.; Girardi, E.; Mounce, B.C.; Weiss, A.; Chane-Woon-Ming, B.; Messmer, M.; Kaukinen, P.; Kopp, A.; Bortolamiol-Becet, D.; Fendri, A.; et al. High-Throughput Fluorescence-Based Screen Identifies the Neuronal MicroRNA miR-124 as a Positive Regulator of Alphavirus Infection. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Kunec, D.; Osterrieder, N. Codon Pair Bias Is a Direct Consequence of Dinucleotide Bias. Cell Rep. 2016, 14, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Butt, A.M.; Nasrullah, I.; Tong, Y. Genome-Wide Analysis of Codon Usage and Influencing Factors in Chikungunya Viruses. PLoS ONE 2014, 9, e90905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Giallonardo, F.; Schlub, T.E.; Shi, M.; Holmes, E.C. Dinucleotide Composition in Animal RNA Viruses Is Shaped More by Virus Family than by Host Species. J. Virol. 2017, 91, e02381-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takata, M.A.; Gonçalves-Carneiro, D.; Zang, T.M.; Soll, S.J.; York, A.; Blanco-Melo, D.; Bieniasz, P.D. CG dinucleotide suppression enables antiviral defence targeting non-self RNA. Nature 2017, 550, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Odon, V.; Fros, J.J.; Goonawardane, N.; Dietrich, I.; Ibrahim, A.; Alshaikhahmed, K.; Nguyen, D.; Simmonds, P. The role of ZAP and OAS3/RNAseL pathways in the attenuation of an RNA virus with elevated frequencies of CpG and UpA dinucleotides. Nucleic Acids Res. 2019, 47, 8061–8083. [Google Scholar] [CrossRef]
- Zhang, Y.; Burke, C.W.; Ryman, K.D.; Klimstra, W.B. Identification and Characterization of Interferon-Induced Proteins That Inhibit Alphavirus Replication. J. Virol. 2007, 81, 11246–11255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bick, M.J.; Carroll, J.-W.N.; Gao, G.; Goff, S.P.; Rice, C.M.; MacDonald, M.R. Expression of the Zinc-Finger Antiviral Protein Inhibits Alphavirus Replication. J. Virol. 2003, 77, 11555–11562. [Google Scholar] [CrossRef] [Green Version]
- Ryman, K.D.; White, L.J.; Johnston, R.E.; Klimstra, W.B. Effects of PKR/RNase L-Dependent and Alternative Antiviral Pathways on Alphavirus Replication and Pathogenesis. Viral Immunol. 2002, 15, 53–76. [Google Scholar] [CrossRef]
- Bréhin, A.-C.; Casadémont, I.; Frenkiel, M.-P.; Julier, C.; Sakuntabhai, A.; Desprès, P. The large form of human 2′,5′-Oligoadenylate Synthetase (OAS3) exerts antiviral effect against Chikungunya virus. Virology 2009, 384, 216–222. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, M.M.H.; Zhao, J.; Li, S.; MacDonald, M.R.; Rice, C.M.; Gao, X.; Gao, G. Sindbis Virus Can Exploit a Host Antiviral Protein To Evade Immune Surveillance. J. Virol. 2016, 90, 10247–10258. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.E.; Long, K.M.; Whitmore, A.C.; Sanders, W.; Thurlow, L.R.; Brown, J.A.; Morrison, C.R.; Vincent, H.; Peck, K.M.; Browning, C.; et al. Disruption of the Opal Stop Codon Attenuates Chikungunya Virus-Induced Arthritis and Pathology. mBio 2017, 8, e01456-17. [Google Scholar] [CrossRef] [Green Version]
- Tuittila, M.T.; Santagati, M.G.; Roytta, M.; Maatta, J.A.; Hinkkanen, A.E. Replicase Complex Genes of Semliki Forest Virus Confer Lethal Neurovirulence. J. Virol. 2000, 74, 4579–4589. [Google Scholar] [CrossRef] [PubMed]
- Myles, K.M.; Kelly, C.L.H.; Ledermann, J.P.; Powers, A.M. Effects of an Opal Termination Codon Preceding the nsP4 Gene Sequence in the O’Nyong-Nyong Virus Genome on Anopheles gambiae Infectivity. J. Virol. 2006, 80, 4992–4997. [Google Scholar] [CrossRef] [Green Version]
- Li, G.P.; Rice, C.M. Mutagenesis of the in-frame opal termination codon preceding nsP4 of Sindbis virus: Studies of translational readthrough and its effect on virus replication. J. Virol. 1989, 63, 1326–1337. [Google Scholar] [CrossRef] [Green Version]
- Firth, A.E.; Wills, N.M.; Gesteland, R.F.; Atkins, J.F. Stimulation of stop codon readthrough: Frequent presence of an extended 3′ RNA structural element. Nucleic Acids Res. 2011, 39, 6679–6691. [Google Scholar] [CrossRef] [Green Version]
- Kendra, J.; Advani, V.M.; Chen, B.; Briggs, J.W.; Zhu, J.; Bress, H.J.; Pathy, S.M.; Dinman, J.D. Functional and structural characterization of the chikungunya virus translational recoding signals. J. Biol. Chem. 2018, 293, 17536–17545. [Google Scholar] [CrossRef] [Green Version]
- Kutchko, K.M.; Madden, E.; Morrison, C.; Plante, K.S.; Sanders, W.; Vincent, H.A.; Cisneros, M.C.C.; Long, K.M.; Moorman, N.J.; Heise, M.T.; et al. Structural divergence creates new functional features in alphavirus genomes. Nucleic Acids Res. 2018, 46, 3657–3670. [Google Scholar] [CrossRef] [Green Version]
- Pichlmair, A.; Schulz, O.; Tan, C.-P.; Rehwinkel, J.; Kato, H.; Takeuchi, O.; Akira, S.; Way, M.; Schiavo, G.; Sousa, C.R.E. Activation of MDA5 Requires Higher-Order RNA Structures Generated during Virus Infection. J. Virol. 2009, 83, 10761–10769. [Google Scholar] [CrossRef] [Green Version]
- Rudd, P.A.; Wilson, J.; Gardner, J.; Larcher, T.; Babarit, C.; Le, T.T.; Anraku, I.; Kumagai, Y.; Loo, Y.-M.; Gale, M.; et al. Interferon Response Factors 3 and 7 Protect against Chikungunya Virus Hemorrhagic Fever and Shock. J. Virol. 2012, 86, 9888–9898. [Google Scholar] [CrossRef] [Green Version]
- Frolova, E.; Frolov, I.; Schlesinger, S. Packaging signals in alphaviruses. J. Virol. 1997, 71, 248–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.Y.; Firth, A.E.; Atasheva, S.; Frolova, E.I.; Frolov, I. Conservation of a Packaging Signal and the Viral Genome RNA Packaging Mechanism in Alphavirus Evolution. J. Virol. 2011, 85, 8022–8036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firth, A.E.; Chung, B.Y.; Fleeton, M.N.; Atkins, J.F. Discovery of frameshifting in Alphavirus 6K resolves a 20-year enigma. Virol. J. 2008, 5, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, J.E.; Kulcsar, K.A.; Schultz, K.L.W.; Riley, C.P.; Neary, J.T.; Marr, S.; Jose, J.; Griffin, D.E.; Kuhn, R.J. Functional Characterization of the Alphavirus TF Protein. J. Virol. 2013, 87, 8511–8523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kendra, J.A.; de la Fuente, C.; Brahms, A.; Woodson, C.; Bell, T.M.; Chen, B.; Khan, Y.A.; Jacobs, J.L.; Kehn-Hall, K.; Dinman, J.D. Ablation of Programmed −1 Ribosomal Frameshifting in Venezuelan Equine Encephalitis Virus Results in Attenuated Neuropathogenicity. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, B.Y.-W.; Firth, A.E.; Atkins, J. Frameshifting in Alphaviruses: A Diversity of 3′ Stimulatory Structures. J. Mol. Biol. 2010, 397, 448–456. [Google Scholar] [CrossRef]
- Frolova, E.I.; Gorchakov, R.; Pereboeva, L.; Atasheva, S.; Frolov, I. Functional Sindbis Virus Replicative Complexes Are Formed at the Plasma Membrane. J. Virol. 2010, 84, 11679–11695. [Google Scholar] [CrossRef] [Green Version]
- Wielgosz, M.M.; Huang, H.V. A novel viral RNA species in Sindbis virus-infected cells. J. Virol. 1997, 71, 9108–9117. [Google Scholar] [CrossRef] [Green Version]
- Levin, J.G.; Friedman, R.M. Analysis of Arbovirus Ribonucleic Acid Forms by Polyacrylamide Gel Electrophoresis. J. Virol. 1971, 7, 504–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruton, C.J.; Kennedy, S.I.T. Semliki Forest Virus Intracellular RNA: Properties of the Multi-stranded RNA Species and Kinetics of Positive and Negative Strand Synthesis. J. Gen. Virol. 1975, 28, 111–127. [Google Scholar] [CrossRef] [PubMed]
- Albulescu, I.C.; Tas, A.; Scholte, F.; Snijder, E.; van Hemert, M. An in vitro assay to study chikungunya virus RNA synthesis and the mode of action of inhibitors. J. Gen. Virol. 2014, 95, 2683–2692. [Google Scholar] [CrossRef] [Green Version]
- Weiss, B.; Goran, D.; Cancedda, R.; Schlesinger, S. Defective Interfering Passages of Sindbis Virus: Nature of the Intracellular Defective Viral RNA. J. Virol. 1974, 14, 1189–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlesinger, S.; Weiss, B.; Dohner, D. Defective particles in alphavirus infections. Med. Biol. 1975, 53, 372–379. [Google Scholar]
- Poirier, E.; Goic, B.; Tomé-Poderti, L.; Frangeul, L.; Boussier, J.; Gausson, V.; Blanc, H.; Vallet, T.; Loyd, H.; Levi, L.I.; et al. Dicer-2-Dependent Generation of Viral DNA from Defective Genomes of RNA Viruses Modulates Antiviral Immunity in Insects. Cell Host Microbe 2018, 23, 353–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, B.; Schlesinger, S. Defective interfering particles of Sindbis virus do not interfere with the homologous virus obtained from persistently infected BHK cells but do interfere with Semliki Forest virus. J. Virol. 1981, 37, 840–844. [Google Scholar] [CrossRef] [Green Version]
- Bruton, C.J.; Porter, A.; Kennedy, S.I.T. Defective-Interfering Particles of Semliki Forest Virus: Intracellular Events During Interference. J. Gen. Virol. 1976, 31, 397–416. [Google Scholar] [CrossRef] [PubMed]
- Levi, L.I.; Rezelj, V.V.; Henrion-Lacritick, A.; Erazo, D.; Boussier, J.; Vallet, T.; Bernhauerová, V.; Suzuki, Y.; Carrau, L.; Weger-Lucarelli, J.; et al. Defective viral genomes from chikungunya virus are broad-spectrum antivirals and prevent virus dissemination in mosquitoes. PLoS Pathog. 2021, 17, e1009110. [Google Scholar] [CrossRef]
- Langsjoen, R.M.; Muruato, A.E.; Kunkel, S.R.; Jaworski, E.; Routh, A. Differential Alphavirus Defective RNA Diversity between Intracellular and Extracellular Compartments Is Driven by Subgenomic Recombination Events. mBio 2020, 11. [Google Scholar] [CrossRef]
- Forrester, N.L.; Guerbois, M.; Adams, A.P.; Liang, X.; Weaver, S.C. Analysis of Intrahost Variation in Venezuelan Equine Encephalitis Virus Reveals Repeated Deletions in the 6-Kilodalton Protein Gene. J. Virol. 2011, 85, 8709–8717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petterson, E.; Stormoen, M.; Evensen, Ø.; Mikalsen, A.B.; Haugland, Ø. Natural infection of Atlantic salmon (Salmo salar L.) with salmonid alphavirus 3 generates numerous viral deletion mutants. J. Gen. Virol. 2013, 94, 1945–1954. [Google Scholar] [CrossRef] [PubMed]
- Portner, A.; Kingsbury, D.W. Homologous Interference by Incomplete Sendai Virus Particles: Changes in Virus-Specific Ribonucleic Acid Synthesis. J. Virol. 1971, 8, 388–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimmock, N.J.; Kennedy, S.I.T. Prevention of Death in Semliki Forest Virus-Infected Mice by Administration of Defective-Interfering Semliki Forest Virus. J. Gen. Virol. 1978, 39, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Barrett, A.D.T.; Dimmock, N.J. Modulation of Semliki Forest Virus-Induced Infection of Mice by Defective-Interfering Virus. J. Infect. Dis. 1984, 150, 98–104. [Google Scholar] [CrossRef]
- Thomson, M.; Dimmock, N. Common Sequence Elements in Structurally Unrelated Genomes of Defective Interfering Semliki Forest Virus. Virology 1994, 199, 366–375. [Google Scholar] [CrossRef]
- Thomson, M.; White, C.L.; Dimmock, N.J. The Genomic Sequence of Defective Interfering Semliki Forest Virus (SFV) Determines Its Ability to Be Replicated in Mouse Brain and to Protect against a Lethal SFV Infectionin Vivo. Virology 1998, 241, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Tapia, K.; Kim, W.-K.; Sun, Y.; Mercado-López, X.; Dunay, E.; Wise, M.; Adu, M.; López, C.B. Defective Viral Genomes Arising In Vivo Provide Critical Danger Signals for the Triggering of Lung Antiviral Immunity. PLoS Pathog. 2013, 9, e1003703. [Google Scholar] [CrossRef] [PubMed]
- Roux, L.; Holland, J.J. Role of defective interfering particles of sendai virus in persistent infections. Virology 1979, 93, 91–103. [Google Scholar] [CrossRef]
- Calain, P.; Monroe, M.C.; Nichol, S.T. Ebola Virus Defective Interfering Particles and Persistent Infection. Virology 1999, 262, 114–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinton, M.A. Characterization of West Nile virus persistent infections in genetically resistant and susceptible mouse cells I. Generation of defective nonplaquing virus particles. Virology 1982, 116, 84–98. [Google Scholar] [CrossRef]
- Ogura, T.; Tanaka, J.; Kamiya, S.; Sato, H.; Ogura, H.; Hatano, M. Human Cytomegalovirus Persistent Infection in a Human Central Nervous System Cell Line: Production of a Variant Virus with Different Growth Characteristics. J. Gen. Virol. 1986, 67, 2605–2616. [Google Scholar] [CrossRef] [PubMed]
- Sekellick, M.J.; Marcus, P.I. Persistent infection I. Interferon-inducing defective-interfering particles as mediators of cell sparing: Possible role in persistent infection by vesicular stomatitis virus. Virology 1978, 85, 175–186. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
LaPointe, A.T.; Sokoloski, K.J. De-Coding the Contributions of the Viral RNAs to Alphaviral Pathogenesis. Pathogens 2021, 10, 771. https://doi.org/10.3390/pathogens10060771
LaPointe AT, Sokoloski KJ. De-Coding the Contributions of the Viral RNAs to Alphaviral Pathogenesis. Pathogens. 2021; 10(6):771. https://doi.org/10.3390/pathogens10060771
Chicago/Turabian StyleLaPointe, Autumn T., and Kevin J. Sokoloski. 2021. "De-Coding the Contributions of the Viral RNAs to Alphaviral Pathogenesis" Pathogens 10, no. 6: 771. https://doi.org/10.3390/pathogens10060771