
Metamorphic Protein Folding Encodes Multiple Anti-Candida Mechanisms in XCL1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
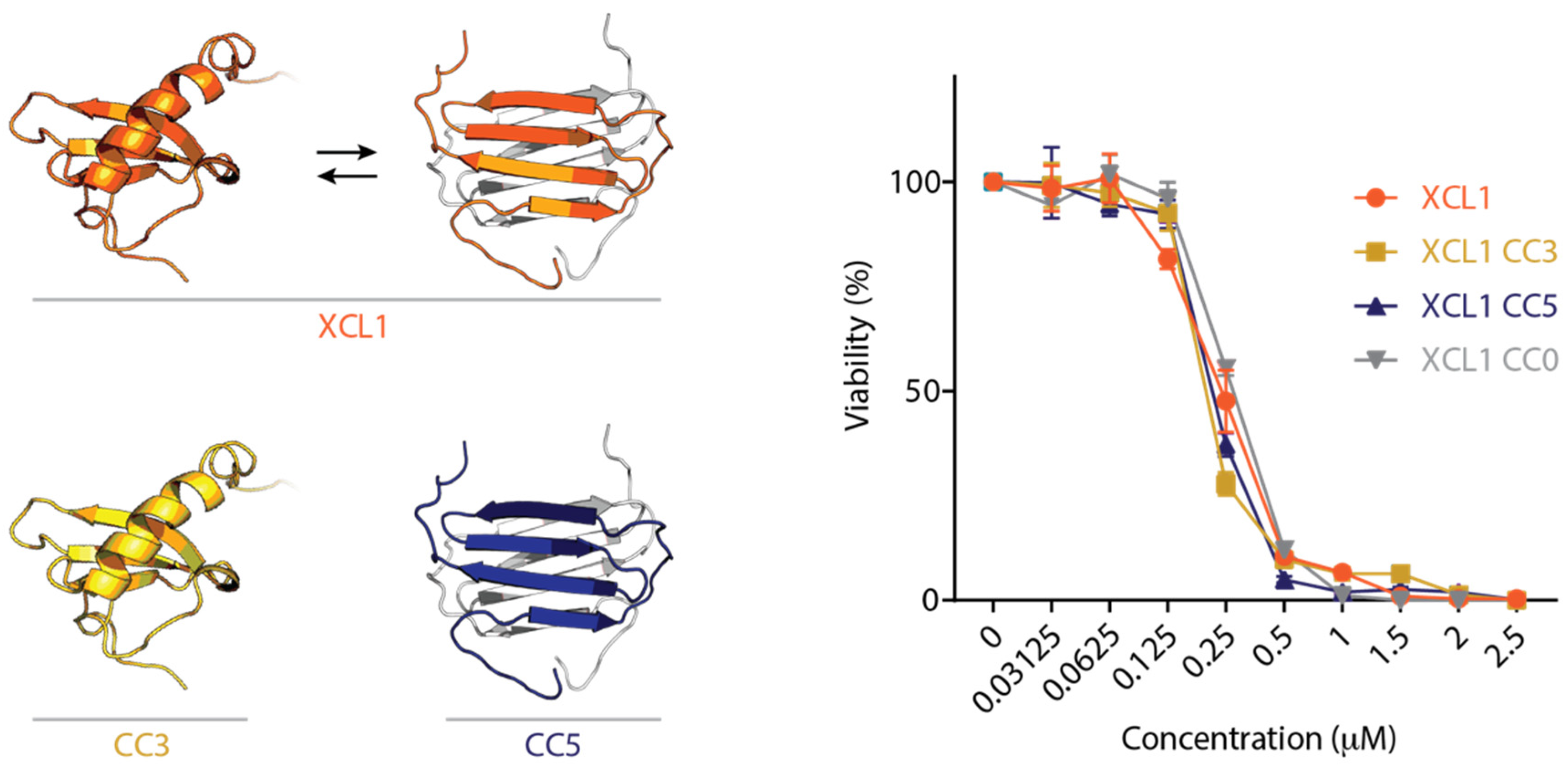
2.1. The XCL1 Chemokine Fold, Alternate Fold, and Unfolded State Kill C. albicans
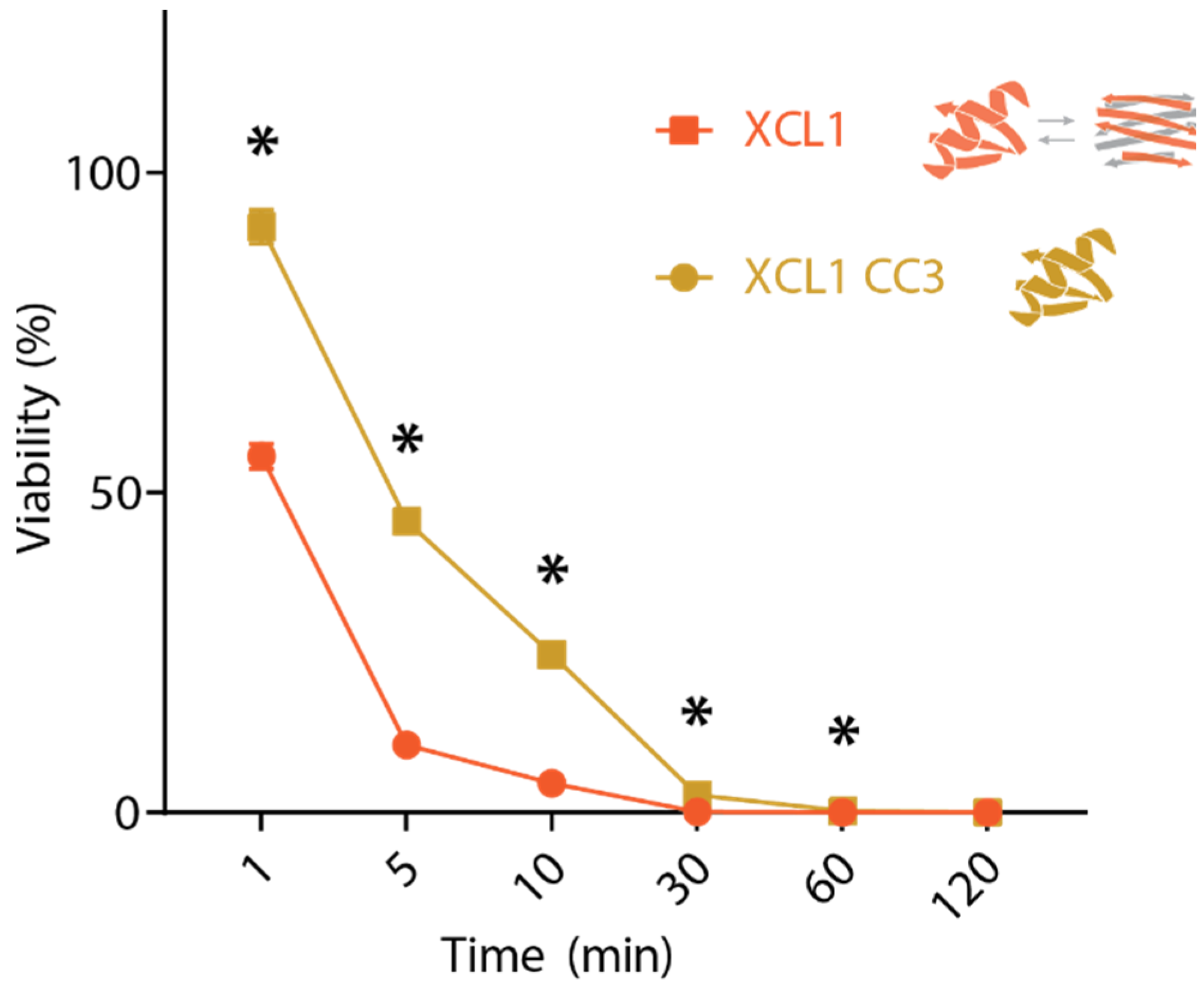
2.2. WT XCL1 Kills C. albicans Faster Than an XCL1 Variant Locked in the Chemokine Fold
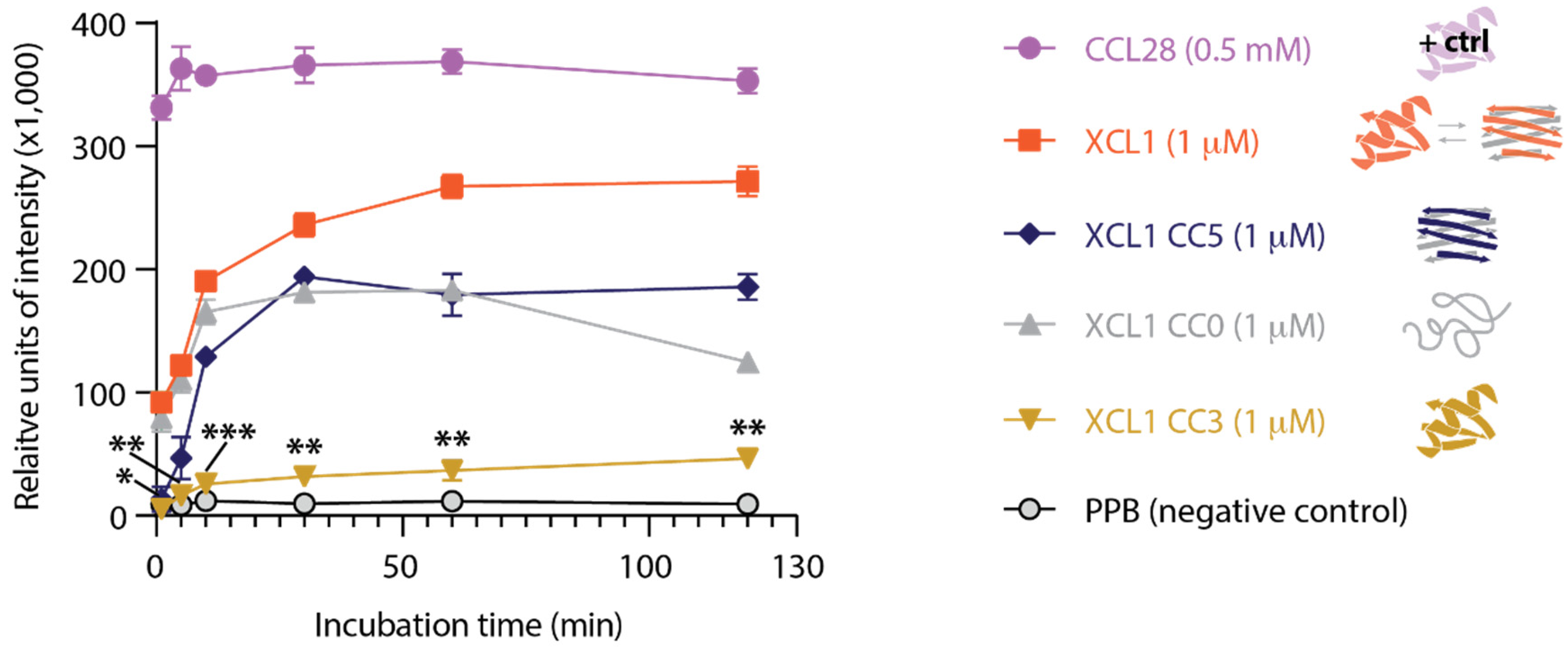
2.3. The XCL1 Alternate Fold Induces More Intense Adenylate Kinase Release from C. albicans Than the XCL1 Chemokine Fold
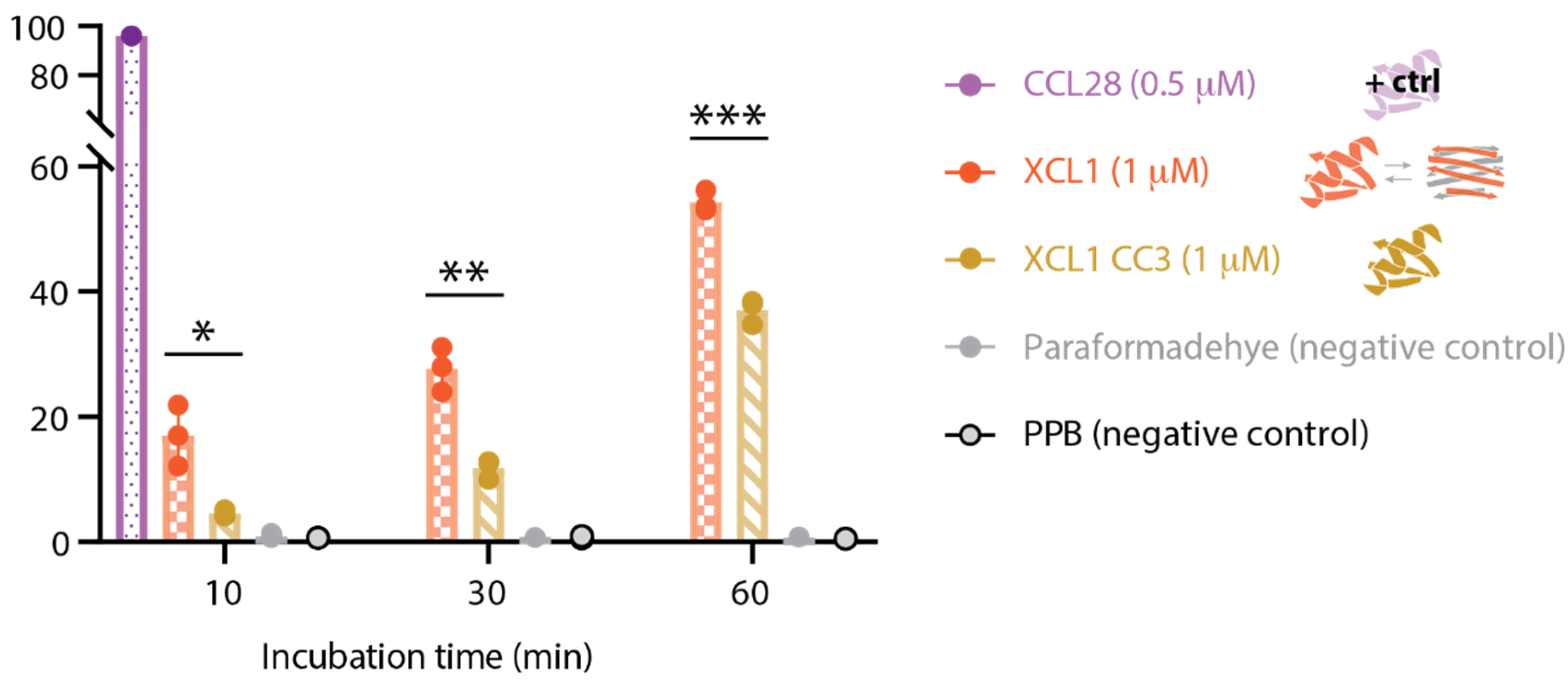
2.4. WT XCL1 Triggers More Propidium Iodide Uptake Than the XCL1 Locked Chemokine Fold Variant
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. Candida Killing Dose-Response Assays
4.3. Adenylate Kinase Release Assays
4.4. Propidium Iodide Uptake Assays
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Webb, B.J.; Ferraro, J.P.; Rea, S.; Kaufusi, S.; Goodman, B.E.; Spalding, J. Epidemiology and Clinical Features of Invasive Fungal Infection in a US Health Care Network. Open Forum Infect. Dis. 2018, 5, ofy187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warris, A.; Pana, Z.D.; Oletto, A.; Lundin, R.; Castagnola, E.; Lehrnbecher, T.; Groll, A.H.; Roilides, E.; Group, E.S. Etiology and Outcome of Candidemia in Neonates and Children in Europe: An 11-year Multinational Retrospective Study. Pediatr. Infect. Dis. J. 2020, 39, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Benedict, K.; Jackson, B.R.; Chiller, T.; Beer, K.D. Estimation of Direct Healthcare Costs of Fungal Diseases in the United States. Clin. Infect. Dis. 2019, 68, 1791–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arastehfar, A.; Gabaldon, T.; Garcia-Rubio, R.; Jenks, J.D.; Hoenigl, M.; Salzer, H.J.F.; Ilkit, M.; Lass-Florl, C.; Perlin, D.S. Drug-Resistant Fungi: An Emerging Challenge Threatening Our Limited Antifungal Armamentarium. Antibiotics 2020, 9, 877. [Google Scholar] [CrossRef]
- Brown, G.D.; Denning, D.W.; Gow, N.A.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci Transl. Med. 2012, 4, 165rv113. [Google Scholar] [CrossRef] [Green Version]
- Basso, V.; Tran, D.Q.; Ouellette, A.J.; Selsted, M.E. Host Defense Peptides as Templates for Antifungal Drug Development. J. Fungi 2020, 6, 241. [Google Scholar] [CrossRef]
- Fernandez de Ullivarri, M.; Arbulu, S.; Garcia-Gutierrez, E.; Cotter, P.D. Antifungal Peptides as Therapeutic Agents. Front. Cell Infect. Microbiol. 2020, 10, 105. [Google Scholar] [CrossRef]
- Dishman, A.F.; Tyler, R.C.; Fox, J.C.; Kleist, A.B.; Prehoda, K.E.; Babu, M.M.; Peterson, F.C.; Volkman, B.F. Evolution of fold switching in a metamorphic protein. Science 2021, 371, 86–90. [Google Scholar] [CrossRef]
- Porter, L.L.; Looger, L.L. Extant fold-switching proteins are widespread. Proc. Natl. Acad. Sci. USA 2018, 115, 5968–5973. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.K.; Porter, L.L. Functional and Regulatory Roles of Fold-Switching Proteins. Structure 2021, 29, 6–14. [Google Scholar] [CrossRef]
- Dishman, A.F.; Volkman, B.F. Unfolding the Mysteries of Protein Metamorphosis. ACS Chem. Biol. 2018, 13, 1438–1446. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Kitahata, K.; Kawabata, F.; Kamei, M.; Hara, Y.; Takamura, S.; Oiso, N.; Kawada, A.; Yoshie, O.; Nakayama, T. A Highly Active Form of XCL1/Lymphotactin Functions as an Effective Adjuvant to Recruit Cross-Presenting Dendritic Cells for Induction of Effector and Memory CD8(+) T Cells. Front. Immunol. 2018, 9, 2775. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Chen, Q.; Hoover, D.M.; Staley, P.; Tucker, K.D.; Lubkowski, J.; Oppenheim, J.J. Many chemokines including CCL20/MIP-3alpha display antimicrobial activity. J. Leukoc Biol. 2003, 74, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Yung, S.C.; Murphy, P.M. Antimicrobial chemokines. Front. Immunol. 2012, 3, 276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuinstra, R.L.; Peterson, F.C.; Kutlesa, S.; Elgin, E.S.; Kron, M.A.; Volkman, B.F. Interconversion between two unrelated protein folds in the lymphotactin native state. Proc. Natl. Acad. Sci. USA 2008, 105, 5057–5062. [Google Scholar] [CrossRef] [Green Version]
- Tuinstra, R.L.; Peterson, F.C.; Elgin, E.S.; Pelzek, A.J.; Volkman, B.F. An engineered second disulfide bond restricts lymphotactin/XCL1 to a chemokine-like conformation with XCR1 agonist activity. Biochemistry 2007, 46, 2564–2573. [Google Scholar] [CrossRef] [Green Version]
- Fox, J.C.; Tyler, R.C.; Guzzo, C.; Tuinstra, R.L.; Peterson, F.C.; Lusso, P.; Volkman, B.F. Engineering Metamorphic Chemokine Lymphotactin/XCL1 into the GAG-Binding, HIV-Inhibitory Dimer Conformation. ACS Chem. Biol. 2015, 10, 2580–2588. [Google Scholar] [CrossRef]
- Dishman, A.F.; Lee, M.W.; de Anda, J.; Lee, E.Y.; He, J.; Huppler, A.R.; Wong, G.C.L.; Volkman, B.F. Switchable Membrane Remodeling and Antifungal Defense by Metamorphic Chemokine XCL1. ACS Infect. Dis. 2020, 6, 1204–1213. [Google Scholar] [CrossRef] [Green Version]
- Nevins, A.M.; Subramanian, A.; Tapia, J.L.; Delgado, D.P.; Tyler, R.C.; Jensen, D.R.; Ouellette, A.J.; Volkman, B.F. A Requirement for Metamorphic Interconversion in the Antimicrobial Activity of Chemokine XCL1. Biochemistry 2016, 55, 3784–3793. [Google Scholar] [CrossRef]
- Thomas, M.A.; He, J.; Peterson, F.C.; Huppler, A.R.; Volkman, B.F. The Solution Structure of CCL28 Reveals Structural Lability that Does Not Constrain Antifungal Activity. J. Mol. Biol. 2018, 430, 3266–3282. [Google Scholar] [CrossRef] [PubMed]
- Hieshima, K.; Ohtani, H.; Shibano, M.; Izawa, D.; Nakayama, T.; Kawasaki, Y.; Shiba, F.; Shiota, M.; Katou, F.; Saito, T.; et al. CCL28 has dual roles in mucosal immunity as a chemokine with broad-spectrum antimicrobial activity. J. Immunol. 2003, 170, 1452–1461. [Google Scholar] [CrossRef] [Green Version]
- Beattie, S.R.; Krysan, D.J. Antifungal drug screening: Thinking outside the box to identify novel antifungal scaffolds. Curr. Opin. Microbiol. 2020, 57, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Krysan, D.J.; Didone, L. A high-throughput screening assay for small molecules that disrupt yeast cell integrity. J. Biomol. Screen 2008, 13, 657–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Thomas, M.A.; de Anda, J.; Lee, M.W.; Van Why, E.; Simpson, P.; Wong, G.C.L.; Grayson, M.H.; Volkman, B.F.; Huppler, A.R. Chemokine CCL28 Is a Potent Therapeutic Agent for Oropharyngeal Candidiasis. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Sun, L.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q. The antimicrobial peptides and their potential clinical applications. Am. J. Transl. Res. 2019, 11, 3919–3931. [Google Scholar]
- Sierra, J.M.; Fuste, E.; Rabanal, F.; Vinuesa, T.; Vinas, M. An overview of antimicrobial peptides and the latest advances in their development. Expert Opin. Biol. Ther. 2017, 17, 663–676. [Google Scholar] [CrossRef]
- Menzies, F.M.; Oldham, R.S.; Waddell, C.; Nelson, S.M.; Nibbs, R.J.B. A Comprehensive Profile of Chemokine Gene Expression in the Tissues of the Female Reproductive Tract in Mice. Immunol. Investig. 2020, 49, 264–286. [Google Scholar] [CrossRef]
- Goodchild, S.C.; Curmi, P.M.G.; Brown, L.J. Structural gymnastics of multifunctional metamorphic proteins. Biophys. Rev. 2011, 3, 143. [Google Scholar] [CrossRef]
- Fox, J.C.; Nakayama, T.; Tyler, R.C.; Sander, T.L.; Yoshie, O.; Volkman, B.F. Structural and agonist properties of XCL2, the other member of the C-chemokine subfamily. Cytokine 2015, 71, 302–311. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dishman, A.F.; He, J.; Volkman, B.F.; Huppler, A.R. Metamorphic Protein Folding Encodes Multiple Anti-Candida Mechanisms in XCL1. Pathogens 2021, 10, 762. https://doi.org/10.3390/pathogens10060762
Dishman AF, He J, Volkman BF, Huppler AR. Metamorphic Protein Folding Encodes Multiple Anti-Candida Mechanisms in XCL1. Pathogens. 2021; 10(6):762. https://doi.org/10.3390/pathogens10060762
Chicago/Turabian StyleDishman, Acacia F., Jie He, Brian F. Volkman, and Anna R. Huppler. 2021. "Metamorphic Protein Folding Encodes Multiple Anti-Candida Mechanisms in XCL1" Pathogens 10, no. 6: 762. https://doi.org/10.3390/pathogens10060762