
Endothelial Dysfunction and SARS-CoV-2 Infection: Association and Therapeutic Strategies

 ,
, 
Abstract
:
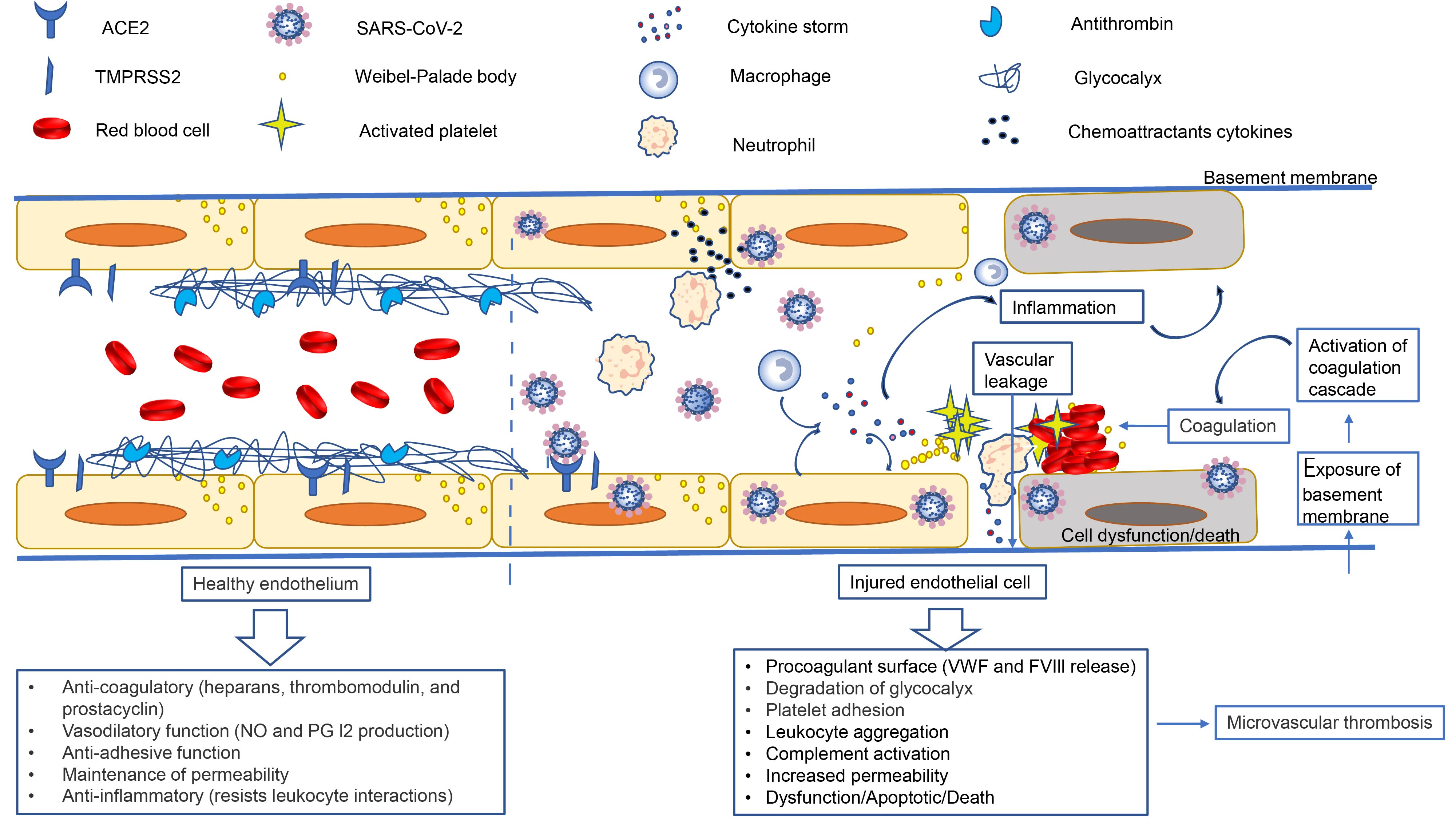{kind=link}
1. Introduction
2. The SARS-CoV-2 Virus
3. Infection of Endothelial Cells by SARS-CoV-2
4. Pathophysiology of Endothelial Dysfunction in COVID-19 Infection
4.1. Endothelial Dysfunction and Immune Responses
4.2. Endothelial Dysfunction and Thrombosis
4.3. Endothelial Dysfunction and Hypercoagulability
4.4. Endothelial Dysfunction and Vascular Permeability
5. Therapeutic Strategies
5.1. Serine Protease Inhibitors
5.2. Renin-Angiotensin–Aldosterone System (RAAS) Inhibitors
5.3. Statins
5.4. Heparin
5.5. Corticosteroids
5.6. Other Agents
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Pons, S.; Fodil, S.; Azoulay, E.; Zafrani, L. The vascular endothelium: The cornerstone of organ dysfunction in severe SARS-CoV-2 infection. Crit. Care 2020, 24, 353. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Miesbach, W.; Makris, M. COVID-19: Coagulopathy, Risk of Thrombosis, and the Rationale for Anticoagulation. Clin. Appl. Thromb. Hemost. 2020, 26, 1076029620938149. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Mao, L.; Jin, H.; Wang, M.; Hu, Y.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D.; et al. Neurologic Manifestations of Hospitalized Patients With Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol. 2020, 77, 683–690. [Google Scholar] [CrossRef] [Green Version]
- Giuffre, M.; Di Bella, S.; Sambataro, G.; Zerbato, V.; Cavallaro, M.; Occhipinti, A.A.; Palermo, A.; Crescenti, A.; Monica, F.; Luzzati, R.; et al. COVID-19-Induced Thrombosis in Patients without Gastrointestinal Symptoms and Elevated Fecal Calprotectin: Hypothesis Regarding Mechanism of Intestinal Damage Associated with COVID-19. Trop. Med. Infect. Dis. 2020, 5, 147. [Google Scholar] [CrossRef]
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032. [Google Scholar] [CrossRef]
- Giuffre, M.; Bozzato, A.M.; Di Bella, S.; Occhipinti, A.A.; Martingano, P.; Cavallaro, M.F.M.; Luzzati, R.; Monica, F.; Cova, M.A.; Croce, L.S. Spontaneous Rectal Perforation in a Patient with SARS-CoV-2 Infection. J. Pers. Med. 2020, 10, 157. [Google Scholar] [CrossRef] [PubMed]
- Clerkin, K.J.; Fried, J.A.; Raikhelkar, J.; Sayer, G.; Griffin, J.M.; Masoumi, A.; Jain, S.S.; Burkhoff, D.; Kumaraiah, D.; Rabbani, L.; et al. COVID-19 and Cardiovascular Disease. Circulation 2020, 141, 1648–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, I.; Limonta, D.; Mahal, L.K.; Hobman, T.C. Endothelium Infection and Dysregulation by SARS-CoV-2: Evidence and Caveats in COVID-19. Viruses 2020, 13, 29. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Levy, J.H.; Levi, M.; Thachil, J. Coagulopathy in COVID-19. J. Thromb. Haemost. 2020, 18, 2103–2109. [Google Scholar] [CrossRef]
- Gavriilaki, E.; Anyfanti, P.; Gavriilaki, M.; Lazaridis, A.; Douma, S.; Gkaliagkousi, E. Endothelial Dysfunction in COVID-19: Lessons Learned from Coronaviruses. Curr. Hypertens. Rep. 2020, 22, 63. [Google Scholar] [CrossRef] [PubMed]
- Chioh, F.W.; Fong, S.W.; Young, B.E.; Wu, K.X.; Siau, A.; Krishnan, S.; Chan, Y.H.; Carissimo, G.; Teo, L.L.; Gao, F.; et al. Convalescent COVID-19 patients are susceptible to endothelial dysfunction due to persistent immune activation. eLife 2021, 10. [Google Scholar] [CrossRef]
- Roumenina, L.T.; Rayes, J.; Frimat, M.; Fremeaux-Bacchi, V. Endothelial cells: Source, barrier, and target of defensive mediators. Immunol. Rev. 2016, 274, 307–329. [Google Scholar] [CrossRef]
- Jin, Y.; Ji, W.; Yang, H.; Chen, S.; Zhang, W.; Duan, G. Endothelial activation and dysfunction in COVID-19: From basic mechanisms to potential therapeutic approaches. Signal. Transduct. Target. Ther. 2020, 5, 293. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Helms, J.; Tacquard, C.; Severac, F.; Leonard-Lorant, I.; Ohana, M.; Delabranche, X.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Fagot Gandet, F.; et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: A multicenter prospective cohort study. Intensive Care Med. 2020, 46, 1089–1098. [Google Scholar] [CrossRef]
- McGonagle, D.; O’Donnell, J.S.; Sharif, K.; Emery, P.; Bridgewood, C. Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia. Lancet Rheumatol. 2020, 2, e437–e445. [Google Scholar] [CrossRef]
- Colling, M.E.; Kanthi, Y. COVID-19-associated coagulopathy: An exploration of mechanisms. Vasc. Med. 2020, 25, 471–478. [Google Scholar] [CrossRef]
- Amraei, R.; Rahimi, N. COVID-19, Renin-Angiotensin System and Endothelial Dysfunction. Cells 2020, 9, 1652. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Becker, R.C. Toward understanding the 2019 Coronavirus and its impact on the heart. J. Thromb. Thrombolysis. 2020, 50, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Campana, P.; Parisi, V.; Leosco, D.; Bencivenga, D.; Della Ragione, F.; Borriello, A. Dendritic Cells and SARS-CoV-2 Infection: Still an Unclarified Connection. Cells 2020, 9, 2046. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Becker, R.C. COVID-19 update: Covid-19-associated coagulopathy. J. Thromb. Thrombolysis. 2020, 50, 54–67. [Google Scholar] [CrossRef]
- Del Turco, S.; Vianello, A.; Ragusa, R.; Caselli, C.; Basta, G. COVID-19 and cardiovascular consequences: Is the endothelial dysfunction the hardest challenge? Thromb. Res. 2020, 196, 143–151. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azizi, S.A.; Azizi, S.A. Neurological injuries in COVID-19 patients: Direct viral invasion or a bystander injury after infection of epithelial/endothelial cells. J. Neurovirol. 2020, 26, 631–641. [Google Scholar] [CrossRef]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- Aimes, R.T.; Zijlstra, A.; Hooper, J.D.; Ogbourne, S.M.; Sit, M.L.; Fuchs, S.; Gotley, D.C.; Quigley, J.P.; Antalis, T.M. Endothelial cell serine proteases expressed during vascular morphogenesis and angiogenesis. Thromb. Haemost. 2003, 89, 561–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishiga, M.; Wang, D.W.; Han, Y.; Lewis, D.B.; Wu, J.C. COVID-19 and cardiovascular disease: From basic mechanisms to clinical perspectives. Nat. Rev. Cardiol. 2020, 17, 543–558. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 183, 1735. [Google Scholar] [CrossRef]
- Stopsack, K.H.; Mucci, L.A.; Antonarakis, E.S.; Nelson, P.S.; Kantoff, P.W. TMPRSS2 and COVID-19: Serendipity or Opportunity for Intervention? Cancer Discov. 2020, 10, 779–782. [Google Scholar] [CrossRef] [Green Version]
- Tu, Y.F.; Chien, C.S.; Yarmishyn, A.A.; Lin, Y.Y.; Luo, Y.H.; Lin, Y.T.; Lai, W.Y.; Yang, D.M.; Chou, S.J.; Yang, Y.P.; et al. A Review of SARS-CoV-2 and the Ongoing Clinical Trials. Int. J. Mol. Sci. 2020, 21, 2657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagele, M.P.; Haubner, B.; Tanner, F.C.; Ruschitzka, F.; Flammer, A.J. Endothelial dysfunction in COVID-19: Current findings and therapeutic implications. Atherosclerosis 2020, 314, 58–62. [Google Scholar] [CrossRef]
- Mason, R.J. Pathogenesis of COVID-19 from a cell biology perspective. Eur. Respir. J. 2020, 55. [Google Scholar] [CrossRef] [Green Version]
- Pober, J.S.; Merola, J.; Liu, R.; Manes, T.D. Antigen Presentation by Vascular Cells. Front. Immunol. 2017, 8, 1907. [Google Scholar] [CrossRef] [Green Version]
- Tamburini, B.A.; Burchill, M.A.; Kedl, R.M. Antigen capture and archiving by lymphatic endothelial cells following vaccination or viral infection. Nat. Commun. 2014, 5, 3989. [Google Scholar] [CrossRef] [PubMed]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Virzi, G.M.; Zhang, J.; Nalesso, F.; Ronco, C.; McCullough, P.A. The Role of Dendritic and Endothelial Cells in Cardiorenal Syndrome. Cardiorenal. Med. 2018, 8, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Betakova, T.; Kostrabova, A.; Lachova, V.; Turianova, L. Cytokines Induced During Influenza Virus Infection. Curr. Pharm. Des. 2017, 23, 2616–2622. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Zimba, O.; Gasparyan, A.Y. Thrombosis in Coronavirus disease 2019 (COVID-19) through the prism of Virchow’s triad. Clin. Rheumatol. 2020, 39, 2529–2543. [Google Scholar] [CrossRef]
- McGonagle, D.; Sharif, K.; O’Regan, A.; Bridgewood, C. The Role of Cytokines including Interleukin-6 in COVID-19 induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun. Rev. 2020, 19, 102537. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Long, X.; Ji, G.; Zhang, B.; Zhang, W.; Zhang, Z.; Chen, X. Clinically significant portal hypertension in cirrhosis patients with COVID-19: Clinical characteristics and outcomes. J. Infect. 2020, 81, e178–e180. [Google Scholar] [CrossRef] [PubMed]
- Desai, T.R.; Leeper, N.J.; Hynes, K.L.; Gewertz, B.L. Interleukin-6 causes endothelial barrier dysfunction via the protein kinase C pathway. J. Surg. Res. 2002, 104, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Laudes, I.J.; Chu, J.C.; Huber-Lang, M.; Guo, R.F.; Riedemann, N.C.; Sarma, J.V.; Mahdi, F.; Murphy, H.S.; Speyer, C.; Lu, K.T.; et al. Expression and function of C5a receptor in mouse microvascular endothelial cells. J. Immunol. 2002, 169, 5962–5970. [Google Scholar] [CrossRef] [Green Version]
- Prendecki, M.; Clarke, C.; Medjeral-Thomas, N.; McAdoo, S.P.; Sandhu, E.; Peters, J.E.; Thomas, D.C.; Willicombe, M.; Botto, M.; Pickering, M.C. Temporal changes in complement activation in haemodialysis patients with COVID-19 as a predictor of disease progression. Clin. Kidney J. 2020, 13, 889–896. [Google Scholar] [CrossRef]
- Merle, N.S.; Noe, R.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part II: Role in Immunity. Front. Immunol. 2015, 6, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricklin, D.; Reis, E.S.; Lambris, J.D. Complement in disease: A defence system turning offensive. Nat. Rev. Nephrol. 2016, 12, 383–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Yuan, X.; Chen, H.; Chaturvedi, S.; Braunstein, E.M.; Brodsky, R.A. Direct activation of the alternative complement pathway by SARS-CoV-2 spike proteins is blocked by factor D inhibition. Blood 2020, 136, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Luscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef]
- Tong, M.; Jiang, Y.; Xia, D.; Xiong, Y.; Zheng, Q.; Chen, F.; Zou, L.; Xiao, W.; Zhu, Y. Elevated Expression of Serum Endothelial Cell Adhesion Molecules in COVID-19 Patients. J. Infect. Dis. 2020, 222, 894–898. [Google Scholar] [CrossRef]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Investig. 2020, 130, 2620–2629. [Google Scholar] [CrossRef] [Green Version]
- Teuwen, L.A.; Geldhof, V.; Pasut, A.; Carmeliet, P. COVID-19: The vasculature unleashed. Nat. Rev. Immunol. 2020, 20, 389–391. [Google Scholar] [CrossRef] [PubMed]
- Kruger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef] [Green Version]
- Jayarangaiah, A.; Kariyanna, P.T.; Chen, X.; Jayarangaiah, A.; Kumar, A. COVID-19-Associated Coagulopathy: An Exacerbated Immunothrombosis Response. Clin. Appl. Thromb. Hemost. 2020, 26, 1076029620943293. [Google Scholar] [CrossRef]
- Steffel, J.; Luscher, T.F.; Tanner, F.C. Tissue factor in cardiovascular diseases: Molecular mechanisms and clinical implications. Circulation 2006, 113, 722–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godo, S.; Shimokawa, H. Endothelial Functions. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e108–e114. [Google Scholar] [CrossRef] [Green Version]
- Iba, T.; Connors, J.M.; Levy, J.H. The coagulopathy, endotheliopathy, and vasculitis of COVID-19. Inflamm. Res. 2020, 69, 1181–1189. [Google Scholar] [CrossRef]
- Wichmann, D. Autopsy Findings and Venous Thromboembolism in Patients with COVID-19. Ann. Intern. Med. 2020, 173, 1030. [Google Scholar] [CrossRef] [PubMed]
- Dolhnikoff, M.; Duarte-Neto, A.N.; de Almeida Monteiro, R.A.; da Silva, L.F.F.; de Oliveira, E.P.; Saldiva, P.H.N.; Mauad, T.; Negri, E.M. Pathological evidence of pulmonary thrombotic phenomena in severe COVID-19. J. Thromb. Haemost. 2020, 18, 1517–1519. [Google Scholar] [CrossRef] [Green Version]
- Loghmani, H.; Conway, E.M. Exploring traditional and nontraditional roles for thrombomodulin. Blood 2018, 132, 148–158. [Google Scholar] [CrossRef] [Green Version]
- Jose, R.J.; Manuel, A. COVID-19 cytokine storm: The interplay between inflammation and coagulation. Lancet Respir Med. 2020, 8, e46–e47. [Google Scholar] [CrossRef]
- Thachil, J. The versatile heparin in COVID-19. J. Thromb. Haemost. 2020, 18, 1020–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Yan, X.; Fan, Q.; Liu, H.; Liu, X.; Liu, Z.; Zhang, Z. D-dimer levels on admission to predict in-hospital mortality in patients with Covid-19. J. Thromb. Haemost. 2020, 18, 1324–1329. [Google Scholar] [CrossRef]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [Green Version]
- Escher, R.; Breakey, N.; Lammle, B. Severe COVID-19 infection associated with endothelial activation. Thromb. Res. 2020, 190, 62. [Google Scholar] [CrossRef] [PubMed]
- Streetley, J.; Fonseca, A.V.; Turner, J.; Kiskin, N.I.; Knipe, L.; Rosenthal, P.B.; Carter, T. Stimulated release of intraluminal vesicles from Weibel-Palade bodies. Blood 2019, 133, 2707–2717. [Google Scholar] [CrossRef] [Green Version]
- Giannotta, M.; Trani, M.; Dejana, E. VE-cadherin and endothelial adherens junctions: Active guardians of vascular integrity. Dev. Cell 2013, 26, 441–454. [Google Scholar] [CrossRef] [Green Version]
- Garvin, M.R.; Alvarez, C.; Miller, J.I.; Prates, E.T.; Walker, A.M.; Amos, B.K.; Mast, A.E.; Justice, A.; Aronow, B.; Jacobson, D. A mechanistic model and therapeutic interventions for COVID-19 involving a RAS-mediated bradykinin storm. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Perico, L.; Benigni, A.; Casiraghi, F.; Ng, L.F.P.; Renia, L.; Remuzzi, G. Immunity, endothelial injury and complement-induced coagulopathy in COVID-19. Nat. Rev. Nephrol. 2021, 17, 46–64. [Google Scholar] [CrossRef] [PubMed]
- Franck, G.; Even, G.; Gautier, A.; Salinas, M.; Loste, A.; Procopio, E.; Gaston, A.T.; Morvan, M.; Dupont, S.; Deschildre, C.; et al. Haemodynamic stress-induced breaches of the arterial intima trigger inflammation and drive atherogenesis. Eur. Heart J. 2019, 40, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Once more unto the breach: Endothelial permeability and atherogenesis. Eur. Heart J. 2019, 40, 938–940. [Google Scholar] [CrossRef]
- Shahin, Y.; Khan, J.A.; Samuel, N.; Chetter, I. Angiotensin converting enzyme inhibitors effect on endothelial dysfunction: A meta-analysis of randomised controlled trials. Atherosclerosis 2011, 216, 7–16. [Google Scholar] [CrossRef]
- Tikoo, K.; Patel, G.; Kumar, S.; Karpe, P.A.; Sanghavi, M.; Malek, V.; Srinivasan, K. Tissue specific up regulation of ACE2 in rabbit model of atherosclerosis by atorvastatin: Role of epigenetic histone modifications. Biochem. Pharmacol. 2015, 93, 343–351. [Google Scholar] [CrossRef]
- Huertas, A.; Montani, D.; Savale, L.; Pichon, J.; Tu, L.; Parent, F.; Guignabert, C.; Humbert, M. Endothelial cell dysfunction: A major player in SARS-CoV-2 infection (COVID-19)? Eur. Respir J. 2020, 56. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhu, Y.; Hong, Y. Decreased Mortality of COVID-19 with Renin-Angiotensin-Aldosterone System Inhibitors Therapy in Patients with Hypertension: A Meta-Analysis. Hypertension 2020, 76, e13–e14. [Google Scholar] [CrossRef]
- Flacco, M.E.; Acuti Martellucci, C.; Bravi, F.; Parruti, G.; Cappadona, R.; Mascitelli, A.; Manfredini, R.; Mantovani, L.G.; Manzoli, L. Treatment with ACE inhibitors or ARBs and risk of severe/lethal COVID-19: A meta-analysis. Heart 2020, 106, 1519–1524. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, H.R.; Adhikari, S.; Pulgarin, C.; Troxel, A.B.; Iturrate, E.; Johnson, S.B.; Hausvater, A.; Newman, J.D.; Berger, J.S.; Bangalore, S.; et al. Renin-Angiotensin-Aldosterone System Inhibitors and Risk of Covid-19. N. Engl. J. Med. 2020, 382, 2441–2448. [Google Scholar] [CrossRef]
- Mancia, G.; Rea, F.; Ludergnani, M.; Apolone, G.; Corrao, G. Renin-Angiotensin-Aldosterone System Blockers and the Risk of Covid-19. N. Engl. J. Med. 2020, 382, 2431–2440. [Google Scholar] [CrossRef] [PubMed]
- Barkas, F.; Milionis, H.; Anastasiou, G.; Liberopoulos, E. Statins and PCSK9 inhibitors: What is their role in coronavirus disease 2019? Med. Hypotheses 2021, 146, 110452. [Google Scholar] [CrossRef] [PubMed]
- Vaduganathan, M.; Vardeny, O.; Michel, T.; McMurray, J.J.V.; Pfeffer, M.A.; Solomon, S.D. Renin-Angiotensin-Aldosterone System Inhibitors in Patients with Covid-19. N. Engl. J. Med. 2020, 382, 1653–1659. [Google Scholar] [CrossRef]
- Suo, Y.; Zhang, Z.; Fu, H.; Zhang, Y.; Yuan, M.; Wang, Y.; Goudis, C.A.; Tse, G.; Liu, T.; Li, G. Inhibition of renin-angiotensin axis reduces the risk of thrombus formation in the left atrial appendage in patients with hypertension complicated by atrial fibrillation. J. Renin Angiotensin Aldosterone Syst. 2018, 19, 1470320318782623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermann, F.; Forster, A.; Chenevard, R.; Enseleit, F.; Hurlimann, D.; Corti, R.; Spieker, L.E.; Frey, D.; Hermann, M.; Riesen, W.; et al. Simvastatin improves endothelial function in patients with rheumatoid arthritis. J. Am. Coll. Cardiol. 2005, 45, 461–464. [Google Scholar] [CrossRef] [Green Version]
- Reriani, M.K.; Dunlay, S.M.; Gupta, B.; West, C.P.; Rihal, C.S.; Lerman, L.O.; Lerman, A. Effects of statins on coronary and peripheral endothelial function in humans: A systematic review and meta-analysis of randomized controlled trials. Eur. J. Cardiovasc. Prev. Rehabil. 2011, 18, 704–716. [Google Scholar] [CrossRef]
- Fedson, D.S. Pandemic influenza: A potential role for statins in treatment and prophylaxis. Clin. Infect. Dis. 2006, 43, 199–205. [Google Scholar] [CrossRef]
- Lee, K.C.H.; Sewa, D.W.; Phua, G.C. Potential role of statins in COVID-19. Int. J. Infect. Dis 2020, 96, 615–617. [Google Scholar] [CrossRef]
- Li, J.; Fan, J.G. Characteristics and Mechanism of Liver Injury in 2019 Coronavirus Disease. J. Clin. Transl. Hepatol. 2020, 8, 13–17. [Google Scholar] [CrossRef] [Green Version]
- Ayerbe, L.; Risco, C.; Ayis, S. The association between treatment with heparin and survival in patients with Covid-19. J. Thromb. Thrombolysis 2020, 50, 298–301. [Google Scholar] [CrossRef]
- Marchandot, B.; Sattler, L.; Jesel, L.; Matsushita, K.; Schini-Kerth, V.; Grunebaum, L.; Morel, O. COVID-19 Related Coagulopathy: A Distinct Entity? J. Clin. Med. 2020, 9, 1651. [Google Scholar] [CrossRef]
- Thachil, J.; Tang, N.; Gando, S.; Falanga, A.; Cattaneo, M.; Levi, M.; Clark, C.; Iba, T. ISTH interim guidance on recognition and management of coagulopathy in COVID-19. J. Thromb. Haemost. 2020, 18, 1023–1026. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, F.; Vitiello, A. Efficacy of synthetic glucocorticoids in COVID-19 endothelites. Naunyn Schmiedebergs Arch. Pharmacol. 2021. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Group, R.C.; Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; et al. Dexamethasone in Hospitalized Patients with Covid-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, H.; Tang, T.-X.; Chen, D.; Tang, L.-S.; Yang, X.-P.; Tang, Z.-H. Endothelial Dysfunction and SARS-CoV-2 Infection: Association and Therapeutic Strategies. Pathogens 2021, 10, 582. https://doi.org/10.3390/pathogens10050582
Deng H, Tang T-X, Chen D, Tang L-S, Yang X-P, Tang Z-H. Endothelial Dysfunction and SARS-CoV-2 Infection: Association and Therapeutic Strategies. Pathogens. 2021; 10(5):582. https://doi.org/10.3390/pathogens10050582
Chicago/Turabian StyleDeng, Hai, Ting-Xuan Tang, Deng Chen, Liang-Sheng Tang, Xiang-Ping Yang, and Zhao-Hui Tang. 2021. "Endothelial Dysfunction and SARS-CoV-2 Infection: Association and Therapeutic Strategies" Pathogens 10, no. 5: 582. https://doi.org/10.3390/pathogens10050582