
Inhibition of Viral Membrane Fusion by Peptides and Approaches to Peptide Design


Abstract
:1. Cellular Entry of Lipid Enveloped Viruses
2. HIV-1
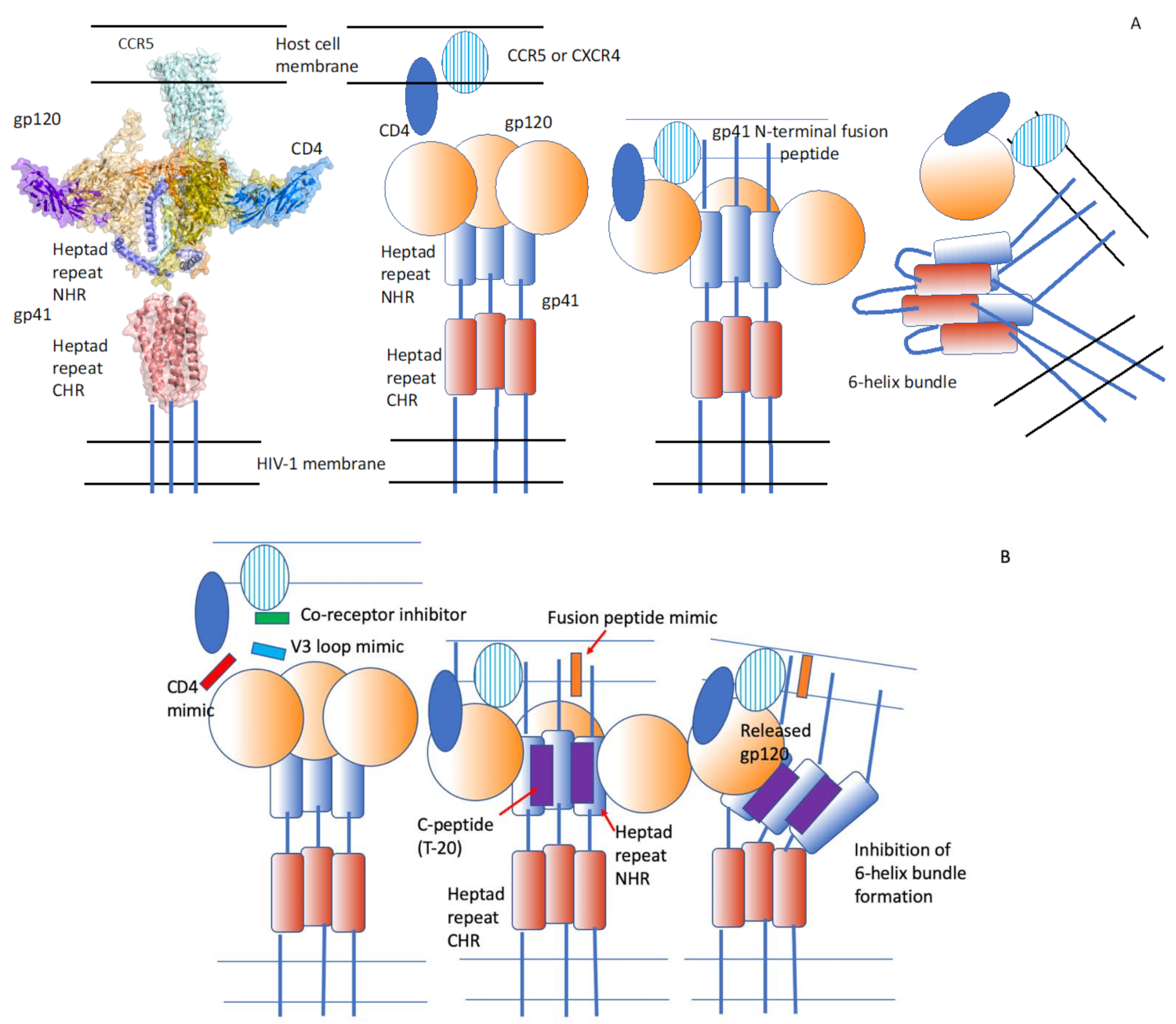
2.1. HIV-1 Infection Is Mediated by the Viral Envelope Glycoprotein (Env) That Attaches to the Cellular Receptor and Induces Virus–Cell Membrane Fusion
2.2. Small Peptides Can Inhibit Membrane Fusion
2.3. CD4- and V3 Loop-Derived Peptides Inhibit Membrane Fusion and Infection
2.4. Peptides Corresponding to the Heptad Repeat Domain of Env Inhibit HIV-1 Membrane Fusion
2.5. Conjugation of Poly(ethylene glycol) to Heptad Repeat Peptides Prolongs Circulation in Blood
2.6. Cholesterol Coupling to Heptad Repeat Peptides Can Enhance Their Fusion Inhibitory Activity
2.7. A Peptide Targeting a Hydrophobic Pocket of HIV-1 gp41 N-terminal Heptad Repeat Trimers Inhibits Infection by Different Virus Isolates
3. Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV)
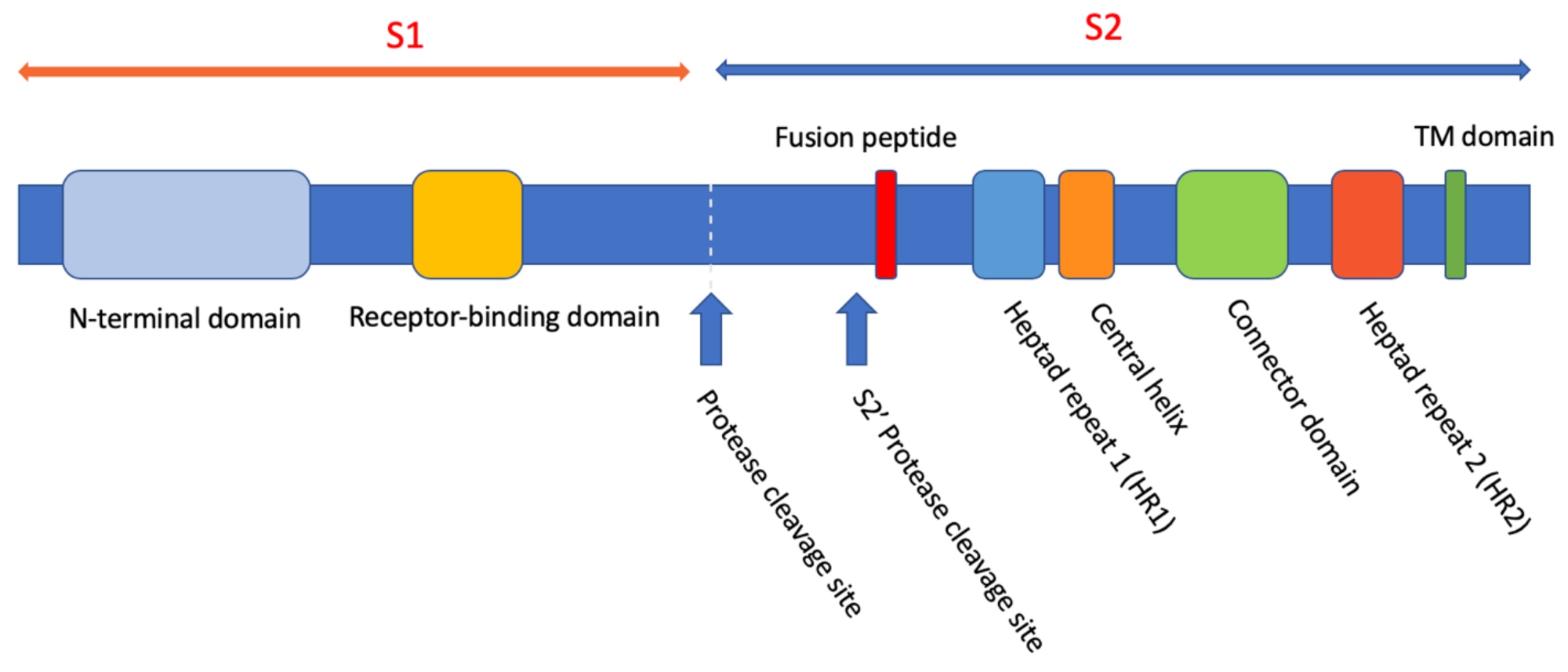
3.1. The C-Terminal S2 Domain of the SARS-CoV Spike Protein S Is Involved in Membrane Fusion with the Host Cell Membrane
3.2. The HR2 Peptide Derived from the C-Terminal Heptad Repeat Region of S Inhibit SARS-CoV Infection
3.3. The Peptides CP-1 and sHR2-8 Derived from the HR2 Region Inhibit Infection at Micromolar Concentrations
3.4. The HR2-Derived Peptides SR9 and SR9EK13 Inhibit Viral Entry at the Cell Surface, following Protease Activation of S at Nanomolar Concentrations
4. Middle East Respiratory Syndrome Coronavirus (MERS-CoV)
4.1. The Peptides HR1P and HR2P Derived from the HR1 and HR2 Regions, Respectively, of the MERS-CoV S2 Protein Form a Six-Helix Bundle, and HR2P Inhibits Membrane Fusion
4.2. The MERS-Five-Helix Bundle, with Three HR1 and Two HR2 Peptides, Inhibits Infection by a Pseudo-Typed Virus
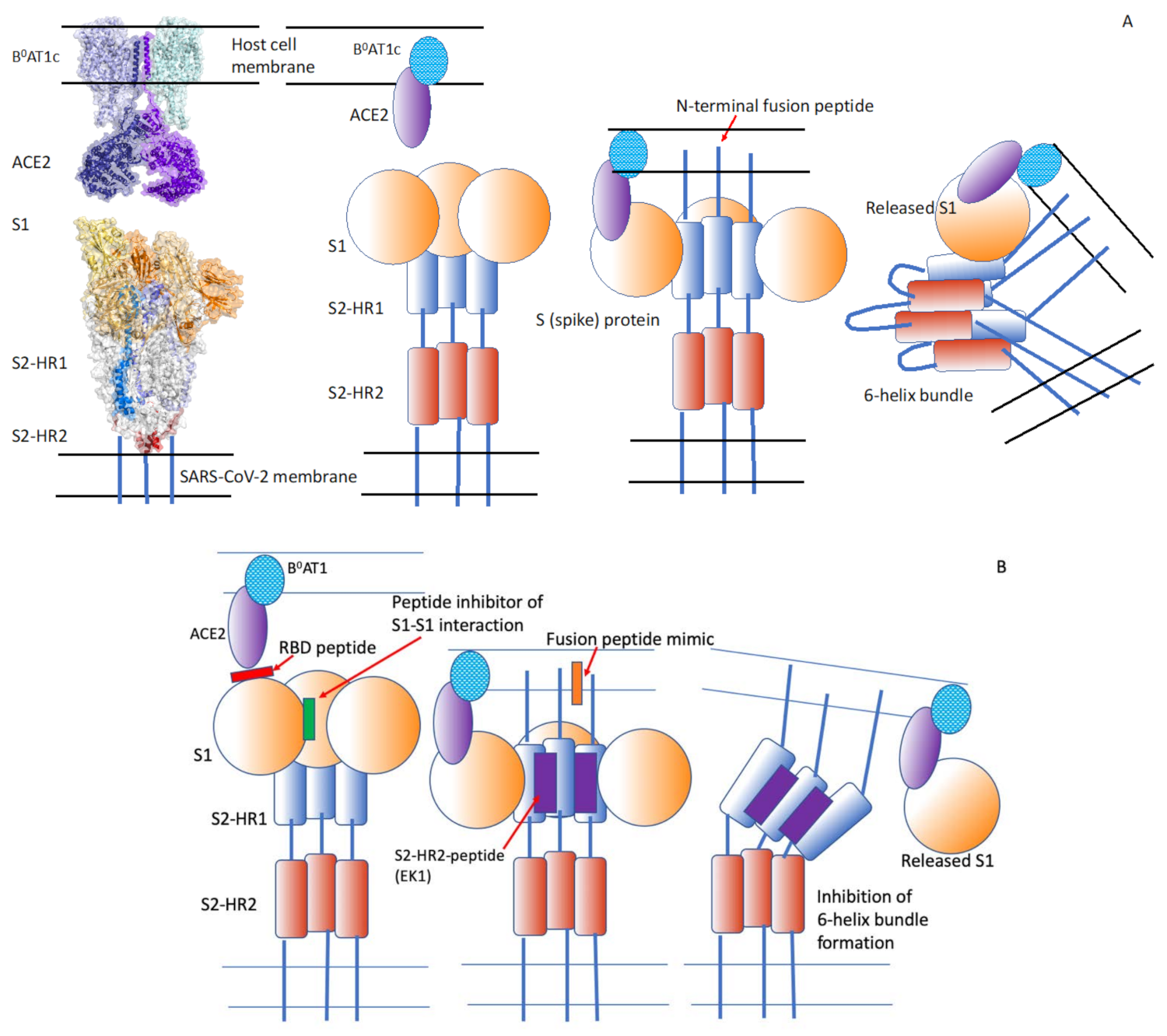
5. Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2)
5.1. The Cholesterol-Coupled, HR1-Targeting Peptides EK1C4 and IPB02 Inhibit Cell–Cell Fusion and Pseudo-Typed Virus Fusion at Nanomolar Concentrations
5.2. There Are Assay- or Cell-Dependent Variations in the Evaluation of Membrane Fusion Activity
6. Influenza Virus
6.1. Influenza Virus Fusion Is Mediated by the Low pH-Induced Conformational Change of the Viral Hemagglutinin
6.2. Influenza Virus Fusion May Be Inhibited by Competitive Inhibitors of the Receptor-Binding Domain of HA
6.3. A Cholesterol-Coupled Peptide Corresponding to a Membrane Proximal Domain of HA Inhibits Influenza Infection at Low Micromolar Concentrations
6.4. A Fusion Inhibitory Peptide Derived from a Helical Region of HA2 Coupled to a Cell-Penetrating Peptide Reduces the Viral Titer in an Animal Model
6.5. The Peptide iHA-100 Generated by a Macrocyclic Peptide Expression System Inhibits Membrane Fusion at Nanomolar Concentrations
6.6. The Complementarity Determining Domains of Neutralizing Antibodies Can Be Used to Design Peptides
7. Hepatitis Viruses
7.1. The Region of the Hepatitis Virus L Protein That Binds to the Cell Surface Sodium-Taurocholate Co-Transporting Polypeptide Receptor has been Identified
7.2. Myrcludex B Inhibits the Binding of Hepatitis B Virus to Its Receptor
7.3. The E2 Protein of Hepatitis C Virus Interacts with Different Cellular Receptor Molecules, including Tetraspanin, Claudin and Transferrin Receptor 1
7.4. Peptides from the E2 Protein and from Claudin-1 Inhibit Infection at Micromolar Concentrations
8. Paramyxovirusess
8.1. The Envelope Protein H Mediates Virion Attachment to the Host Cell Membrane, and the F Protein Carries out the Membrane Fusion Reaction That Involves the Anti-Parallel Alignment of Heptad Repeat Domains
8.2. A Short Peptide Analogous to the N-Terminus of the Cleaved F Protein Can Inhibit Sendai Virus Fusion
8.3. The Peptides HRC4 and HRC2 Derived from the Measles Virus F-Protein C-Terminal Heptad Repeat Region Inhibit Measles Virus Infection at Nanomolar Concentrations
9. Flaviviruses
9.1. The Class II E Protein of Flaviviruses Binds to Phosphatidylserine Receptors on Host Cells, and its Low pH-Induced Conformational Change in Endosomes Mediates the Interaction between Two Domains of the Protein
9.2. Peptides Derived from the E Protein Stem Helix 2 Inhibit Infection of Japanese Encephalitis Virus at Nanomolar IC50
10. Herpesviruses
10.1. Herpesviruses Utilize a Complex Set of Membrane Proteins to Eventually Activate the gB Protein, which Mediates Membrane Fusion by Inserting into the Host Cell Membrane and Changing Conformation
10.2. Peptides It1b and MelN4 Designed to Have Membrane “Interfacial Activity” Inhibit HSV-1 Infection at Low Micromolar Concentrations
10.3. The Peptide gBh1m Corresponding to the N-terminal Domain of the Coiled-Coil Structure of gB and Modified with Poly(ethylene glycol) and Cholesterol Inhibits HSV-1 Infection
11. Filoviruses
11.1. The Ebola Virus Membrane Protein GP1 Is Cleaved in Endosomes and the Cleaved Form GPcl Binds the Cholesterol Transporter Niemann-Pick C1
11.2. Conjugation of the HIV-1 Tat Peptide to an Ebola Virus CHR Peptide Mediates Endosome Localization and Enhances Antiviral Activity
11.3. Peptides Can Have Inhibitory Effects across Virus Families
11.4. Cyclic Peptides That Bind GPcl Can Inhibit Infection by a Pseudotyped Virus
12. Protein Design Methods to Target Protein Interfaces
13. Sequence-Based Methods
13.1. Relying on the Primary Sequence of Proteins, Synthesized Peptides Can Mimic the Key Contacts between Interacting Proteins and Inhibit Their Normal Functions
13.2. Large-Scale Peptide Screening, Utilizing Phage-Display, mRNA or DNA-Encoded Libraries, Can Generate Inhibitory Molecules following Large-Scale Screening
14. Structure-Based Methods
14.1. The Precise Structural Information of the Target Protein or Protein Complex Can Be Used in Modeling and Designing Inhibitory Peptides
14.2. Chemical Modifications of the Peptides, including Carbon-Stapling and Peptide Cyclization, Can Preserve Their Structural Integrity
15. PiPRED: A Knowledge- and Structure-Based Method
15.1. The Natural Behavior of Peptide Sequences Can Be Utilized to Design Bioactive Peptides
15.2. The Entire Peptide–Protein Interface Is Systematically Explored during Modeling in a Knowledge-Based Approach to Find the Optimal Conformations
16. PePI-Covid19 Database: A Repertoire of Peptides to Target SARS-CoV-2 Binding to ACE2
16.1. Macromolecules such as the Soluble ACE2 Receptor for SARS-CoV-2, Computationally Modified ACE2, Mini-Proteins and Aptamers Can Inhibit Viral Infection
16.2. The PePI-Covid19 Database Was Established Based on the PiPreD Method to Design Peptides That Target the Receptor-Binding Region of the S Protein
17. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Strauss, J.H.; Strauss, E.G. Viruses and Human Disease; Elsevier: Amsterdam, The Netherlands; Academic Press: Burlington, NJ, USA; San Diego, CA, USA; London, UK, 2008; p. vii+468. [Google Scholar]
- Düzgüneş, N. Medical Microbiology and Immunology for Dentistry; Quintessence Publishing: Chicago, IL, USA, 2016; p. ix+293. [Google Scholar]
- Schibli, D.J.; Weissenhorn, W. Class I and class II viral fusion protein structures reveal similar principles in membrane fusion. Mol. Membr. Biol. 2004, 21, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Weissenhorn, W.; Hinz, A.; Gaudin, Y. Virus membrane fusion. FEBS Lett. 2007, 581, 2150–2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backovic, M.; Jardetzky, T.S. Class III viral membrane fusion proteins. Curr. Opin. Struct. Biol. 2009, 19, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limburg, H.; Harbig, A.; Bestle, D.; Stein, D.A.; Moulton, H.M.; Jaeger, J.; Janga, H.; Hardes, K.; Koepke, J.; Schulte, L.; et al. TMPRSS2 is the major activating protease of influenza A virus in primary human airway cells and influenza B virus in human type II pneumocytes. J. Virol. 2019, 93, e00649-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Copeland, C.S.; Doms, R.W.; Bolzau, E.M.; Webster, R.G.; Helenius, A. Assembly of influenza hemagglutinin trimers and its role in intracellular transport. J. Cell Biol. 1986, 103, 1179–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinz, F.X.; Allison, S.L. The machinery for flavivirus fusion with host cell membranes. Curr. Opin. Microbiol. 2001, 4, 450–455. [Google Scholar] [CrossRef]
- Stiasny, K.; Heinz, F.X. Flavivirus membrane fusion. J. Gen. Virol. 2006, 87, 2755–2766. [Google Scholar] [CrossRef]
- Kielian, M.; Rey, F.A. Virus membrane-fusion proteins: More than one way to make a hairpin. Nat. Rev. Microbiol. 2006, 4, 67–76. [Google Scholar] [CrossRef]
- Modis, Y. Class II fusion proteins. Adv. Exp. Med. Biol. 2013, 790, 150–166. [Google Scholar]
- Baquero, E.; Albertini, A.A.; Gaudin, Y. Recent mechanistic and structural insights on class III viral fusion glycoproteins. Curr. Opin. Struct. Biol. 2015, 33, 52–60. [Google Scholar] [CrossRef]
- Nicola, A.V. Herpesvirus entry into host cells mediated by endosomal low pH. Traffic 2016, 17, 965–975. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Finzi, A.; Sodroski, J. The conformational states of the HIV-1 envelope glycoproteins. Trends Microbiol. 2020, 28, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Lu, M.; Gorman, J.; Terry, D.S.; Hong, X.; Zhou, Z.; Zhao, H.; Altman, R.B.; Arthos, J.; Blanchard, S.C.; et al. HIV-1 Env trimer opens through an asymmetric intermediate in which individual protomers adopt distinct conformations. eLife 2018, 7, e34271. [Google Scholar] [CrossRef] [PubMed]
- Howard, A.R.; Munro, J.B. Developments in single-molecule and single-particle fluorescence-based approaches for studying viral envelope glycoprotein dynamics and membrane fusion. Adv. Virus Res. 2019, 104, 123–146. [Google Scholar]
- Richardson, C.D.; Scheid, A.; Choppin, P.W. Specific inhibition of paramyxovirus and myxovirus replication by oligopeptides with amino acid sequences similar to those at the N-termini of the F1 or HA2 viral polypeptides. Virology 1980, 105, 205–222. [Google Scholar] [CrossRef]
- Konopka, K.; Pretzer, E.; Düzgüneş, N. Differential effects of a hydrophobic tripeptide on human immunodeficiency virus type 1 (HIV-1)-induced syncytium formation and viral infectivity. Biochem. Biophys. Res. Commun. 1995, 208, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Owens, R.J.; Tanner, C.C.; Mulligan, M.J.; Srinivas, R.V.; Compans, R.W. Oligopeptide inhibitors of HIV-induced syncytium formation. AIDS Res. Hum. Retrovir. 1990, 1990 6, 1289–1296. [Google Scholar] [CrossRef]
- Nara, P.L.; Hwang, K.M.; Rausch, D.M.; Lifson, J.D.; Eiden, L.E. CD4 antigen-based antireceptor peptides inhibit infectivity of human immunodeficiency virus in vitro at multiple stages of the viral life cycle. Proc. Natl. Acad. Sci. USA 1989, 86, 7139–7143. [Google Scholar] [CrossRef] [Green Version]
- Slepushkin, V.A.; Salem, I.I.; Andreev, S.M.; Dazin, P.; Düzgüneş, N. Targeting of liposomes to HIV-1-infected cells by peptides derived from the CD4 receptor. Biochem. Biophys. Res. Commun. 1996, 227, 827–833. [Google Scholar] [CrossRef]
- Benjouad, A.; Chapuis, F.; Fenouillet, E.; Gluckman, J.C. Multibranched peptide constructs derived from the V3 loop of envelope glycoprotein gp120 inhibit human immunodeficiency virus type 1 infection through interaction with CD4. Virology 1995, 206, 457–464. [Google Scholar] [CrossRef]
- Wild, C.T.; Shugars, D.C.; Greenwell, T.K.; McDanal, C.B.; Matthews, T.J. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. USA 1994, 91, 9770–9774. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.H.; Matthews, T.J.; McDanal, C.B.; Bolognesi, D.P.; Greenberg, M.L. A molecular clasp in the human immunodeficiency virus (HIV) type 1 TM protein determines the anti-HIV activity of gp41 derivatives: Implication for viral fusion. J. Virol. 1995, 69, 3771–3777. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Cheng, S.; Zhang, Y.; Ding, Y.; Chong, H.; Xing, H.; Jiang, S.; Li, X.; Ma, L. long-acting HIV-1 fusion inhibitory peptides and their mechanisms of action. Viruses 2019, 11, 811. [Google Scholar] [CrossRef] [Green Version]
- Ingallinella, P.; Bianchi, E.; Ladwa, N.A.; Wang, Y.-J.; Hrin, R.; Veneziano, M.; Bonelli, F.; Ketas, T.J.; Moore, J.P.; Miller, M.D.; et al. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc. Natl. Acad. Sci. USA 2009, 106, 5801–5806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welch, B.D.; Francis, J.N.; Redman, J.S.; Paul, S.; Weinstock, M.T.; Reeves, J.D.; Lie, Y.S.; Whitby, F.G.; Eckert, D.M.; Hill, C.P. Design of a potent D-peptide HIV-1 entry inhibitor with a strong barrier to resistance. J. Virol. 2010, 84, 11235–11244. [Google Scholar] [CrossRef] [Green Version]
- Parry, D.A. Coiled-coils in alpha-helix-containing proteins: Analysis of the residue types within the heptad repeat and the use of these data in the prediction of coiled-coils in other proteins. Biosci. Rep. 1982, 1982 2, 1017–1024. [Google Scholar] [CrossRef]
- Xu, Y.; Lou, Z.; Liu, Y.; Pang, H.; Tien, P.; Gao, G.F.; Rao, Z. Crystal structure of severe acute respiratory syndrome coronavirus spike protein fusion core. J. Biol. Chem. 2004, 279, 49414–49419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Debnath, A.K. Development of HIV entry inhibitors targeted to the coiled-coil regions of gp41. Biochem. Biophys. Res. Commun. 2000, 269, 641–646. [Google Scholar] [CrossRef]
- Kilby, J.M.; Hopkins, S.; Venetta, T.M.; DiMassimo, B.; Cloud, G.A.; Lee, J.Y.; Alldredge, L.; Hunter, E.; Lambert, D.; Bolognesi, D.; et al. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat. Med. 1998, 4, 1302–1307. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.; Fass, D.; Berger, J.M.; Kim, P.S. Core structure of gp41 from the HIV envelope glycoprotein. Cell 1997, 89, 263–273. [Google Scholar] [CrossRef] [Green Version]
- Gallo, S.A.; Puri, A.; Blumenthal, R. HIV-1 gp41 six-helix bundle formation occurs rapidly after the engagement of gp120 by CXCR4 in the HIV-1 Env-mediated fusion process. Biochemistry 2001, 40, 12231–12236. [Google Scholar] [CrossRef]
- Liu, S.; Lu, H.; Niu, J.; Xu, Y.; Wu, S.; Jiang, S. Different from the HIV fusion inhibitor C34, the anti-HIV drug Fuzeon (T-20) inhibits HIV-1 entry by targeting multiple sites in gp41 and gp120. J. Biol. Chem. 2005, 280, 11259–11273. [Google Scholar] [CrossRef] [Green Version]
- Welch, B.D.; VanDemark, A.P.; Heroux, A.; Hill, C.P.; Kay, M.S. Potent D-peptide inhibitors of HIV-1 entry. Proc. Natl. Acad. Sci. USA 2007, 104, 16828–16833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belouzard, S.; Millet, J.K.; Licitra, B.N.; Whittaker, G.R. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 2012, 4, 1011–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Xiao, G.; Chen, Y.; He, Y.; Niu, J.; Escalante, C.R.; Xiong, H.; Farmar, J.; Debnath, A.K.; Tien, P.; et al. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: Implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet 2004, 363, 938–947. [Google Scholar] [CrossRef] [Green Version]
- Kliger, Y.; Levanon, E.Y. Cloaked similarity between HIV-1 and SARS-CoV suggests an anti-SARS strategy. BMC Microbiol. 2003, 3, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Xiao, G.; Xu, Y.; Yuan, F.; Zheng, C.; Liu, Y.; Yan, H.; Cole, D.K.; Bell, J.I.; Rao, Z.; et al. Following the rule: Formation of the 6-helix bundle of the fusion core from severe acute respiratory syndrome coronavirus spike protein and identification of potent peptide inhibitors. Biochem. Biophys. Res. Commun. 2004, 319, 283–288. [Google Scholar] [CrossRef]
- Yuan, K.; Yi, L.; Chen, J.; Qu, X.; Qing, T.; Rao, X.; Jiang, P.; Hu, J.; Xiong, Z.; Nie, Y.; et al. Suppression of SARS-CoV entry by peptides corresponding to heptad regions on spike glycoprotein. Biochem. Biophys. Res. Commun. 2004, 319, 746–752. [Google Scholar] [CrossRef]
- Bosch, B.J.; Martina, B.E.E.; van der Zee, R.; Lepault, J.; Haijema, B.J.; Versluis, C.; Heck, A.J.R.; de Groot, R.; Osterhaus, A.D.M.E.; Rottier, P.J.M. Severe acute respiratory syndrome coronavirus (SARS-CoV) infection inhibition using spike protein heptad repeat-derived peptides. Proc. Natl. Acad. Sci. USA 2004, 101, 8455–8460. [Google Scholar] [CrossRef] [Green Version]
- Liu, I.J.; Kao, C.L.; Hsieh, S.C.; Wey, M.T.; Kan, L.S.; Wang, W.K. Identification of a minimal peptide derived from heptad repeat (HR) 2 of spike protein of SARS-CoV and combination of HR1-derived peptides as fusion inhibitors. Antivir. Res. 2009, 81, 82–87. [Google Scholar] [CrossRef]
- Ujike, M.; Nishikawa, H.; Otaka, A.; Yamamoto, N.; Yamamoto, N.; Matsuoka, M.; Kodama, E.; Fujii, N.; Taguchi, F. Heptad repeat-derived peptides block protease-mediated direct entry from the cell surface of severe acute respiratory syndrome coronavirus but not entry via the endosomal pathway. J. Virol. 2008, 82, 588–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raj, V.S.; Mou, H.; Smits, S.L.; Dekkers, D.H.; Müller, M.A.; Dijkman, R.; Muth, D.; Demmers, J.A.; Zaki, A.; Fouchier, R.A.; et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 2013, 495, 251–254. [Google Scholar] [CrossRef] [Green Version]
- Barlan, A.; Zhao, J.; Sarkar, M.K.; Li, K.; McCray, P.B., Jr.; Perlman, S.; Gallagher, T. Receptor variation and susceptibility to Middle East respiratory syndrome coronavirus infection. J. Virol. 2014, 88, 4953–4961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Liu, Q.; Zhu, Y.; Chan, K.H.; Qin, L.; Li, Y.; Wang, Q.; Chan, J.F.; Du, L.; Yu, F.; et al. Structure-based discovery of Middle East respiratory syndrome coronavirus fusion inhibitor. Nat. Commun. 2014, 5, 3067. [Google Scholar] [CrossRef] [Green Version]
- Channappanavar, R.; Lu, L.; Xia, S.; Du, L.; Meyerholz, D.K.; Perlman, S.; Jiang, S. Protective effect of intranasal regimens containing peptidic Middle East respiratory syndrome coronavirus fusion inhibitor against MERS-CoV infection. J. Infect. Dis. 2015, 212, 1894–1903. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, H.; Shi, J.; Zhang, Z.; Gong, R. Identification of a novel inhibitor against Middle East respiratory syndrome coronavirus. Viruses 2017, 9, 255. [Google Scholar] [CrossRef] [Green Version]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Outlaw, V.K.; Bovier, F.T.; Mears, M.C.; Cajimat, M.N.; Zhu, Y.; Lin, M.J.; Addetia, A.; Lieberman, N.A.P.; Peddu, V.; Xie, X.; et al. Inhibition of coronavirus entry in vitro and ex vivo by a lipid-conjugated peptide derived from the SARS-CoV-2 spike glycoprotein HRC domain. mBio 2020, 11, e01935-20. [Google Scholar] [CrossRef]
- Zhu, Y.; Yu, D.; Yan, H.; Chong, H.; He, Y. Design of potent membrane fusion inhibitors against SARS-CoV-2, an emerging coronavirus with high fusogenic activity. J. Virol. 2020, 94, e00635-20. [Google Scholar] [CrossRef]
- Bertram, S.; Glowacka, I.; Steffen, I.; Kühl, A.; Pöhlmann, S. Novel insights into proteolytic cleavage of influenza virus hemagglutinin. Rev. Med. Virol. 2010, 20, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, B.S.; Whittaker, G.R. Cleavage activation of human-adapted influenza virus subtypes by kallikrein-related peptidases 5 and 12. J. Biol. Chem. 2013, 288, 17399–17407. [Google Scholar] [CrossRef] [Green Version]
- White, J.M.; Wilson, I.A. Anti-peptide antibodies detect steps in a protein conformational change: Low-pH activation of the influenza virus hemagglutinin. J. Cell Biol. 1987, 105, 2887–2896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stegmann, T.; Booy, F.P.; Wilschut, J. Effects of low pH on influenza virus. Activation and inactivation of the membrane fusion capacity of the hemagglutinin. J. Biol. Chem. 1987, 262, 17744–17749. [Google Scholar] [CrossRef]
- Düzgüneş, N.; Gambale, F. Membrane action of synthetic N-terminal peptides of influenza virus hemagglutinin and its mutants. FEBS Lett. 1988, 227, 110–114. [Google Scholar] [CrossRef] [Green Version]
- Düzgüneş, N.; Shavnin, S.A. Membrane destabilization by N-terminal peptides of viral envelope proteins. J. Membr. Biol. 1992, 128, 71–80. [Google Scholar] [CrossRef]
- Düzgüneş, N.; Pedroso de Lima, M.C.; Stamatatos, L.; Flasher, D.; Alford, D.; Friend, D.S.; Nir, S. Fusion activity and inactivation of influenza virus: Kinetics of low pH-induced fusion with cultured cells. J. Gen. Virol. 1992, 73, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Ramalho-Santos, J.; Nir, S.; Düzgüneş, N.; Pato de Carvalho, A.; Pedroso de Lima, M.C. A common mechanism for influenza virus fusion activity and inactivation. Biochemistry 1993, 32, 2771–2779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsubara, T.; Onishi, A.; Saito, T.; Shimada, A.; Inoue, H.; Taki, T.; Nagata, K.; Okahata, Y.; Sato, T. Sialic acid-mimic peptides as hemagglutinin inhibitors for anti-influenza therapy. J. Med. Chem. 2010, 53, 4441–4449. [Google Scholar] [CrossRef]
- Jones, J.C.; Turpin, E.A.; Bultmann., H.; Brandt, C.R.; Schultz-Cherry, S. Inhibition of influenza virus infection by a novel antiviral peptide that targets viral attachment to cells. J. Virol. 2006, 80, 11960–11967. [Google Scholar] [CrossRef] [Green Version]
- Perrier, A.; Eluard, M.; Petitjean, M.; Vanet, A. In silico design of new inhibitors against hemagglutinin of influenza. J. Phys. Chem. B 2019, 123, 582–592. [Google Scholar] [CrossRef]
- Lee, K.K.; Pessi, A.; Gui, L.; Santoprete, A.; Talekar, A.; Moscona, A.; Porotto, M. Capturing a fusion intermediate of influenza hemagglutinin with a cholesterol-conjugated peptide, a new antiviral strategy for influenza virus. J. Biol. Chem. 2011, 286, 42141–42149. [Google Scholar] [CrossRef] [Green Version]
- Figueira, T.N.; Augusto, M.T.; Rybkina, K.; Stelitano, D.; Noval, M.G.; Harder, O.E.; Veiga, A.S.; Huey, D.; Alabi, C.A.; Biswas, S.; et al. Effective in vivo targeting of influenza virus through a cell-penetrating/fusion inhibitor tandem peptide anchored to the plasma membrane. Bioconjug. Chem. 2018, 29, 3362–3376. [Google Scholar] [CrossRef]
- Hipolito, C.J.; Suga, H. Ribosomal production and in vitro selection of natural product-like peptidomimetics: The FIT and RaPID systems. Curr. Opin. Chem. Biol. 2012, 16, 196–203. [Google Scholar] [CrossRef]
- Saito, M.; Itoh, Y.; Yasui, F.; Munakata, T.; Yamane, D.; Ozawa, M.; Ito, R.; Katoh, T.; Ishigaki, H.; Nakayama, M. Macrocyclic peptides exhibit antiviral effects against influenza virus HA and prevent pneumonia in animal models. Nat. Commun. 2021, 12, 2654. [Google Scholar] [CrossRef]
- Kadam, R.U.; Juraszek, J.; Brandenburg, B.; Buyck, C.; Schepens, W.B.G.; Kesteleyn, B.; Stoops, B.; Vreeken, R.J.; Vermond, J.; Goutier, W.; et al. Potent peptidic fusion inhibitors of influenza virus. Science 2017, 358, 496–502. [Google Scholar] [CrossRef] [Green Version]
- Glebe, D.; Urban, S.; Knoop, E.V.; Çaǧ, N.; Krass, P.; Grün, S.; Bulavaite, A.; Sasnauskas, K.; Gerlich, W.H. Mapping of the hepatitis B virus attachment site by use of infection-inhibiting preS1 lipopeptides and Tupaia hepatocytes. Gastroenterology 2005, 129, 234–245. [Google Scholar] [CrossRef]
- Persing, D.H.; Varmus, H.E.; Ganem, D. The pre-S1 protein of hepatitis B virus is acylated at its amino terminus with myristic acid. J. Virol. 1987, 61, 1672–1677. [Google Scholar] [CrossRef] [Green Version]
- Gripon, P.; Le Seyec, J.; Rumin, S.; Guguen-Guillouzo, C. Myristylation of the hepatitis B virus large surface protein is essential for viral infectivity. Virology 1995, 213, 292–299. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012, 1, e00049. [Google Scholar] [CrossRef] [PubMed]
- Gripon, P.; Rumin, S.; Urban, S.; Le Seyec, J.; Glaise, D.; Cannie, I.; Guyomard, C.; Lucas, J.; Trepo, C.; Guguen-Guillouzo, C. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. USA 2002, 99, 15655–15660. [Google Scholar] [CrossRef] [Green Version]
- Gripon, P.; Cannie, I.; Urban, S. Efficient inhibition of hepatitis B virus infection by acylated peptides derived from the large viral surface protein. J. Virol. 2005, 79, 1613–1622. [Google Scholar] [CrossRef] [Green Version]
- Petersen, J.; Dandri, M.; Mier, W.; Lütgehetmann, M.; Volz, T.; von Weizsäcker, F.; Haberkorn, U.; Fischer, L.; Pollok, J.-M.; Erbes, B.; et al. Prevention of hepatitis B virus infection in vivo by entry inhibitors derived from the large envelope protein. Nat. Biotechnol. 2008, 26, 335–341. [Google Scholar] [CrossRef]
- Meier, A.; Mehrle, S.; Weiss, T.S.; Mier, W.; Urban, S. Myristoylated PreS1-domain of the hepatitis B virus L-protein mediates specific binding to differentiated hepatocytes. Hepatology 2013, 58, 31–42. [Google Scholar] [CrossRef]
- Volz, T.; Allweiss, L.; Ben, M.; Barek, M.; Warlich, M.; Lohse, A.W.; Pollok, J.M.; Alexandrov, A.; Urban, S.; Peterson, J.; et al. The entry inhibitor myrcludex- B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with Hepatitis B Virus. J. Hepatol. 2013, 58, 861–867. [Google Scholar] [CrossRef]
- Deterding, K.; Wedemeyer, H. Beyond pegylated interferon-alpha: New treatments for hepatitis delta. AIDS Rev. 2019, 21, 126–134. [Google Scholar] [CrossRef]
- LeBlanc, E.V.; Kim, Y.; Capicciotti, C.J.; Colpitts, C.C. Hepatitis C virus glycan-dependent interactions and the potential for novel preventative strategies. Pathogens 2021, 10, 685. [Google Scholar] [CrossRef]
- Si, Y.; Liu, S.; Liu, X.; Jacobs, J.L.; Cheng, M.; Niu, Y.; Jin, Q.; Wang, T.; Yang, W. A human claudin-1-derived peptide inhibits hepatitis C virus entry. Hepatology 2012, 56, 507–515. [Google Scholar] [CrossRef] [Green Version]
- Lindenbach, B.D.; Rice, C.M. The ins and outs of hepatitis C virus entry and assembly. Nat. Rev. Microbiol. 2013, 11, 688–700. [Google Scholar] [CrossRef] [Green Version]
- Zeisel, M.B.; Fofana, I.; Fafi-Kremer, S.; Baumert, T.F. Hepatitis C virus entry into hepatocytes: Molecular mechanisms and targets for antiviral therapies. Review. J. Hepatol. 2011, 54, 566–576. [Google Scholar] [CrossRef]
- Zeisel, M.B.; Felmlee, D.J.; Baumert, T.F. Hepatitis C virus entry. Review. Curr. Top. Microbiol. Immunol. 2013, 369, 87–112. [Google Scholar]
- Zhu, Y.Z.; Qian, X.J.; Zhao, P.; Qi, Z.T. How hepatitis C virus invades hepatocytes: The mystery of viral entry. World J. Gastroenterol. 2014, 20, 3457–3467. [Google Scholar] [CrossRef] [PubMed]
- Alhammad, Y.M.; Maharajh, S.; Butcher, R.; Eden, J.S.; White, P.A.; Poumbourios, P.; Drummer, H.E. Longitudinal sequence and functional evolution within glycoprotein E2 in hepatitis C virus genotype 3a infection. PLoS ONE 2015, 10, e0126397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douan, F.; Lavellette, D.; Cosset, F.-L. The mechanism of HCV entry into host cells. Prog. Mol. Biol. Transl. Sci. 2015, 129, 63–106. [Google Scholar]
- Chang, C.-C.; Hsu, H.-J.; Yen, J.-H.; Lo, S.-Y.; Liou, J.-W. A sequence in the loop domain of hepatitis C virus E2 protein identified in silico as crucial for the selective binding to human CD81. PLoS ONE 2017, 12, e0177383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrensch, F.; Crouchet, E.; Ligat, G.; Zeisel, M.B.; Keck, Z.-Y.; Foung, S.K.H.; Schuster, C.; Baumert, T.F. Hepatitis C virus (HCV)-apolipoprotein interactions and immune evasion and their impact on HCV vaccine design. Front. Immunol. 2018, 9, 1436. [Google Scholar] [CrossRef] [PubMed]
- Colpitts, C.C.; Tsai, P.-L.; Zeisel, M.B. Hepatitis C virus entry: An intriguingly complex and highly regulated process. Int. J. Mol. Sci. 2020, 21, 2091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabahi, A.; Uprichard, S.L.; Wimley, W.C.; Dash, S.; Garry, R.F. Unexpected structural of features of the hepatitis C virus envelope protein 2 ectodomain. J. Virol. 2014, 88, 10280–10288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, X.; Niu, Y.; Cheng, M.; Liu, X.; Feng, Y.; Zheng, F.; Fan, J.; Li, X.; Jin, Q.; Zhong, J.; et al. Identification of a potent and broad-spectrum hepatitis C virus fusion inhibitory peptide from the E2 stem domain. Sci. Rep. 2016, 6, 25224. [Google Scholar] [CrossRef] [Green Version]
- Moss, W.J.; Griffin, D.E. Global measles elimination. Nat. Rev. Microbiol. 2006, 4, 900–908. [Google Scholar] [CrossRef]
- Azarm, K.D.; Lee, B. Differential features of fusion activation within the Paramyxoviridae. Viruses 2020, 12, 161. [Google Scholar] [CrossRef] [Green Version]
- Dutch, R.E.; Jardetzky, T.S.; Lamb, R.A. Virus membrane fusion proteins: Biological machines that undergo a metamorphosis. Biosci. Rep. 2000, 20, 597–612. [Google Scholar] [CrossRef]
- Baker, K.A.; Dutch, R.E.; Lamb, R.A.; Jardetzky, T.S. Structural basis for paramyxovirus-mediated membrane fusion. Mol. Cell. 1999, 3, 309–319. [Google Scholar] [CrossRef]
- Mathieu, C.; Augusto, M.T.; Niewiesk, S.; Horvat, B.; Palermo, L.M.; Sanna, G.; Maddedu, S.; Huey, D.; Castanho, M.A.R.B.; Porotto, M.; et al. Broad spectrum antiviral activity for paramyxoviruses is modulated by biophysical properties of fusion inhibitory peptides. Sci. Rep. 2017, 7, 43610. [Google Scholar] [CrossRef] [Green Version]
- Kelsey, D.R.; Flanagan, T.D.; Young, J.; Yeagle, P.L. Peptide inhibitors of enveloped virus infection inhibit phospholipid vesicle fusion and Sendai virus fusion with phospholipid vesicles. J. Biol. Chem. 1990, 265, 12178–12183. [Google Scholar] [CrossRef]
- Epand, R.M. Virus replication inhibitory peptide inhibits the conversion of phospholipid bilayers to the hexagonal phase. Biosci. Rep. 1986, 6, 647–653. [Google Scholar] [CrossRef]
- Rapaport, D.; Ovadia, M.; Shai, Y. A synthetic peptide corresponding to a conserved heptad repeat domain is a potent inhibitor of Sendai virus-cell fusion: An emerging similarity with functional domains of other viruses. EMBO J. 1995, 14, 5524–5531. [Google Scholar] [CrossRef]
- Lambert, D.M.; Barney, S.; Lambert, A.L.; Guthrie, K.; Medinas, R.; Davis, D.E.; Bucy, T.; Erickson, J.; Merutka, G.; Petteway, S.R., Jr. Peptides from conserved regions of paramyxovirus fusion (F) proteins are potent inhibitors of viral fusion. Proc. Natl. Acad. Sci. USA 1996, 93, 2186–2191. [Google Scholar] [CrossRef] [Green Version]
- Yao, Q.; Compans, R.W. Peptides corresponding to the heptad repeat sequence of human parainfluenza virus fusion protein are potent inhibitors of virus infection. Virology 1996, 223, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Welsch, J.C.; Talekar, A.; Mathieu, C.; Pessi, A.; Moscona, A.; Horvat, B.; Porotto, M. Fatal measles virus infection prevented by brain-penetrant fusion inhibitors. J. Virol. 2013, 87, 13785–13794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Düzgüneş, N.; Konopka, K. Peptide inhibitors of viral membrane fusion. Med. Res. Arch. 2020, 8, 1–33. [Google Scholar] [CrossRef]
- Figueira, T.N.; Palermo, L.M.; Veiga, A.S.; Huey, D.; Alabi, A.; Santos, N.C.; Welsch, J.C.; Mathieum, C.; Horvat, B.; Niewiesk, S.; et al. In vivo efficacy of measles virus fusion protein-derived peptides is modulated by the properties of self-assembly and membrane residence. J. Virol. 2016, 91, e01554-16. [Google Scholar] [CrossRef] [Green Version]
- Porotto, M.; Yokoyama, C.C.; Palermo, L.M.; Mungall, B.; Aljofan, M.; Cortese, R.; Pessi, A.; Moscona, A. Viral entry inhibitors targeted to the membrane site of action. J. Virol. 2010, 84, 6760–6768. [Google Scholar] [CrossRef] [Green Version]
- Laureti, M.; Narayanan, D.; Rodriguez-Andres, J.; Fazakerley, J.K.; Kedzierski, L. Flavivirus receptors: Diversity, identity, and cell entry. Front. Immunol. 2018, 9, 2180. [Google Scholar] [CrossRef] [Green Version]
- Meertens, L.; Carnec, X.; Lecoin, M.P.; Ramdasi, R.; Guivel-Benhassine, F.; Lew, E.; Lemke, G.; Schwartz, O.; Amara, A. The TIM and TAM families of phosphatidylserine receptors mediate dengue virus entry. Cell Host Microbe 2012, 12, 544–557. [Google Scholar] [CrossRef] [Green Version]
- Perera-Lecoin, M.; Meertens, L.; Carnec, X.; Amara, A. Flavivirus entry receptors: An update. Viruses 2013, 6, 69–88. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, A.G.; Yang, P.L.; Harrison, S.C. Peptide inhibitors of dengue-virus entry target a late-stage fusion intermediate. PLoS Pathog. 2010, 6, e1000851. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Ohyama, A. Association between the pH-dependent conformational change of West Nile flavivirus E protein and virus-mediated membrane fusion. J. Gen. Virol. 1988, 69, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Christian, E.A.; Kahle, K.M.; Mattia, K.; Puffer, B.A.; Pfaff, J.M.; Miller, A.; Paes, C.; Davidson, E.; Doranz, B.J. Atomic-level functional model of dengue virus envelope protein infectivity. Proc. Natl. Acad. Sci. USA 2013, 110, 18662–18667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Liu, Y.; Wang, S.; Sun, J.; Wang, P.; Xin, Q.; Zhang, L.; Xiao, G.; Wang, W. Antiviral activity of peptide inhibitors derived from the protein E stem against Japanese encephalitis and Zika viruses. Antivir. Res. 2017, 141, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Costin, J.M.; Jenwitheesuk, E.; Lok, S.M.; Hunsperger, E.; Conrads, K.A.; Fontaine, K.A.; Rees, C.R.; Rossman, M.G.; Isern, S.; Samudrala, R.; et al. Structural optimization and de novo design of dengue virus entry inhibitory peptides. PLoS Negl. Trop. Dis. 2010, 4, e721. [Google Scholar] [CrossRef] [PubMed]
- Connolly, S.A.; Jardetzky, T.S.; Longnecker, R. The structural basis of herpesvirus entry. Nat. Rev. Microbiol. 2021, 19, 110–121. [Google Scholar] [CrossRef]
- Vollmer, B.; Grünewald, K. Herpesvirus membrane fusion—A team effort. Curr. Opin. Struct. Biol. 2020, 62, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, L.; Falanga, A.; Del Genio, V.; Palomba, L.; Galdiero, M.; Franci, G.; Galdiero, S. A boost to the antiviral activity: Cholesterol tagged peptides derived from glycoprotein B of Herpes Simplex virus type I. Int. J. Biol. Macromol. 2020, 162, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Atanasiu, D.; Saw, W.T.; Cohen, G.H.; Eisenberg, R.J. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J. Virol. 2010, 84, 12292–12299. [Google Scholar] [CrossRef] [Green Version]
- Gianni, T.; Menotti, L.; Campadelli-Fiume, G. A heptad repeat in herpes simplex virus 1 gH, located downstream of the alpha-helix with attributes of a fusion peptide, is critical for virus entry and fusion. J. Virol. 2005, 79, 7042–7049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianni, T.; Piccoli, A.; Bertucci, C.; Campadelli-Fiume, G. Heptad repeat 2 in herpes simplex virus 1 gH interacts with heptad repeat 1 and is critical for virus entry and fusion. J. Virol. 2006, 80, 2216–2224. [Google Scholar] [CrossRef] [Green Version]
- Galdiero, S.; Vitiello, M.; D’Isanto, M.; Falanga, A.; Collins, C.; Raieta, K.; Pedone, C.; Browne, H.; Galdiero, M. Analysis of synthetic peptides from heptad-repeat domains of herpes simplex virus type 1 glycoproteins H and B. J. Gen. Virol. 2006, 87, 1085–1097. [Google Scholar] [CrossRef]
- Galdiero, S.; Falanga, A.; Vitiello, M.; D’Isanto, M.; Cantisani, M.; Kampanaraki, A.; Benedetti, E.; Browne, H.; Galdiero, M. Peptides containing membrane-interacting motifs inhibit herpes simplex virus type 1 infectivity. Peptides 2008, 29, 1461–1471. [Google Scholar] [CrossRef]
- Hoffmann, A.R.; Guha, S.; Wu, E.; Ghimire, J.; Wang, Y.; He, J.; Garry, R.F.; Wimley, W.C. Broad-spectrum antiviral entry inhibition by interfacially active peptides. J. Virol. 2020, 94, e01682-20. [Google Scholar] [CrossRef]
- Vollmer, B.; Pražák, V.; Vasishtan, D.; Jefferys, E.E.; Hernandez-Duran, A.; Vallbracht, M.; Klupp, B.G.; Mettenleiter, T.C.; Backovic, M.; Rey, F.A.; et al. The prefusion structure of herpes simplex virus glycoprotein B. Sci. Adv. 2020, 6, eabc1726. [Google Scholar] [CrossRef]
- Miller, E.H.; Chandran, K. Filovirus entry into cells—New insights. Curr. Opin. Virol. 2012, 2, 206–214. [Google Scholar] [CrossRef] [Green Version]
- Fénéant, L.; Szymańska-de Wijs, K.M.; Nelson, E.A.; White, J.M. An exploration of conditions proposed to trigger the Ebola virus glycoprotein for fusion. PLoS ONE 2019, 14, e0219312. [Google Scholar] [CrossRef]
- Jain, S.; Martynova, E.; Rizvanov, A.; Khaiboullina, S.; Baranwal, M. Structural and functional aspects of Ebola virus proteins. Pathogens 2021, 10, 1330. [Google Scholar] [CrossRef]
- Markosyan, R.M.; Miao, C.; Zheng, Y.M.; Melikyan, G.B.; Liu, S.L.; Cohen, F.S. Induction of cell-cell fusion by Ebola virus glycoprotein: Low pH is not a trigger. PLoS Pathog. 2016, 12, e1005373. [Google Scholar] [CrossRef]
- Miller, E.H.; Harrison, J.S.; Radoshitzky, S.R.; Higgins, C.D.; Chi, X.; Dong, L.; Kuhn, J.H.; Bavari, S.; Lai, J.R.; Chandran, K. Inhibition of Ebola virus entry by a C-peptide targeted to endosomes. J. Biol. Chem. 2011, 286, 15854–15861. [Google Scholar] [CrossRef] [Green Version]
- Koehler, J.W.; Smith, J.M.; Ripoll, D.R.; Spik, K.W.; Taylor, S.L.; Badger, C.V.; Grant, R.J.; Ogg, M.M.; Wallqvist, A.; Guttieri, M.C.; et al. A fusion-inhibiting peptide against Rift Valley fever virus inhibits multiple, diverse viruses. PLoS Negl. Trop. Dis. 2013, 7, e2430. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Ma, L.; Yi, D.; Wang, H.; Wang, J.; Zhang, Y.; Guo, Y.; Li, X.; Zhou, J.; Shi, Y.; et al. Novel cyclo-peptides inhibit Ebola pseudotyped virus entry by targeting primed GP protein. Antivir. Res. 2018, 155, 1–11. [Google Scholar] [CrossRef]
- Petsalaki, E.; Russell, R.B. Peptide-mediated interactions in biological systems: New discoveries and applications. Curr. Opin. Biotechnol. 2008, 19, 344–350. [Google Scholar] [CrossRef]
- Valkov, E.; Sharpe, T.; Marsh, M.; Greive, S.; Hyvonen, M. Targeting protein-protein interactions and fragment-based drug discovery. Top. Curr. Chem. 2012, 317, 145–179. [Google Scholar]
- Mullard, A. Protein-protein interaction inhibitors get into the groove. Nat. Rev. Drug Discov. 2012, 11, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Oliva, B.; Fernandez-Fuentes, N. Knowledge-based modeling of peptides at protein interfaces: PiPreD. Bioinformatics 2015, 31, 1405–1410. [Google Scholar] [CrossRef] [Green Version]
- Molina, R.; Oliva, B.; Fernandez-Fuentes, N. A collection of designed peptides to target SARS-Cov-2-ACE2 interaction: PepI-Covid19 database. bioRxiv 2020. [Google Scholar] [CrossRef]
- Mack, E.; Ziv, E.; Reuveni, H.; Kalman, R.; Niv, M.Y.; Jorns, A.; Lenzen, S.; Shafrir, E. Prevention of insulin resistance and beta-cell loss by abrogating PKCepsilon-induced serine phosphorylation of muscle IRS-1 in Psammomys obesus. Diabetes. Metab. Res. Rev. 2008, 24, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Wexler, I.D.; Niv, M.Y.; Reuveni, H. Sequence-based protein kinase inhibition: Applications for drug development. BioTechniques 2005, 39, S575–S576. [Google Scholar] [CrossRef]
- Yeon, S.W.; Jeon, Y.J.; Hwang, E.M.; Kim, T.Y. Effects of peptides derived from BACE1 catalytic domain on APP processing. Peptides 2007, 28, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Muratspahic, E.; Freissmuth, M.; Gruber, C.W. Nature-derived peptides: A growing niche for GPCR ligand discovery. Trends Pharmacol. Sci. 2019, 40, 309–326. [Google Scholar] [CrossRef]
- Sudarman, E.; Bollati-Fogolín, M.; Hafner, M.; Müller, W.; Scheller, J.; Rose-John, S.; Eichler, J. Synthetic mimetics of the gp130 binding site for viral interleukin-6 as inhibitors of the vIL-6–gp130 Interaction. Chem. Biol. Drug Des. 2008, 71, 494–500. [Google Scholar] [CrossRef]
- Goodman, S.L.; Holzemann, G.; Sulyok, G.A.; Kessler, H. Nanomolar small molecule inhibitors for αvβ6, αvβ5, and αvβ3 integrins. J. Med. Chem. 2002, 45, 1045–1051. [Google Scholar] [CrossRef]
- Rubinstein, M.; Niv, M.Y. Peptidic modulators of protein-protein interactions: Progress and challenges in computational design. Biopolymers 2009, 91, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Jenny-Avital, E.R. Enfuvirtide, an HIV-1 fusion inhibitor. N. Engl. J. Med. 2003, 349, 1770–1771. [Google Scholar]
- Garcia-Garcia, J.; Valls-Comamala, V.; Guney, E.; Andreu, D.; Munoz, F.J.; Fernandez-Fuentes, N.; Oliva, B. iFrag: A protein-protein interface prediction server based on sequence fragments. J. Mol. Biol. 2017, 429, 382–389. [Google Scholar] [CrossRef]
- Clackson, T.; Wells, J.A. In vitro selection from protein and peptide libraries. Trends Biotechnol. 1994, 12, 173–184. [Google Scholar] [CrossRef]
- Wilson, D.S.; Keefe, A.D.; Szostak, J.W. The use of mRNA display to select high-affinity protein-binding peptides. Proc. Natl. Acad. Sci. USA 2001, 98, 3750–3755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, G.; Huang, Y.; Zhou, Y.; Li, Y.; Li, X. Future challenges with DNA-encoded chemical libraries in the drug discovery domain. Expert Opin. Drug Discov. 2019, 14, 735–753. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.C.; Pan, X.; Yang, H.; Wan, L.K.; Stewart-Jones, G.; Dorrell, L.; Ogg, G. Linking genotype to phenotype on beads: High throughput selection of peptides with biological function. Sci. Rep. 2013, 3, 3030. [Google Scholar] [CrossRef] [Green Version]
- Lam, K.S.; Lebl, M.; Krchnak, V. The “one-bead-one-compound” combinatorial library method. Chem. Rev. 1997, 97, 411–448. [Google Scholar] [CrossRef]
- Szymczak, L.C.; Kuo, H.Y.; Mrksich, M. Peptide arrays: Development and application. Anal. Chem. 2018, 90, 266–282. [Google Scholar] [CrossRef]
- Quartararo, A.J.; Gates, Z.P.; Somsen, B.A.; Hartrampf, N.; Ye, X.; Shimada, A.; Kajihara, Y.; Ottmann, C.; Pentelute, B.L. Ultra-large chemical libraries for the discovery of high-affinity peptide binders. Nat. Comm. 2020, 11, 3183. [Google Scholar] [CrossRef]
- Stevers, L.M.; Sijbesma, E.; Botta, M.; MacKintosh, C.; Obsil, T.; Landrieu, I.; Cau, Y.; Wilson, A.J.; Karawajczyk, A.; Eickhoff, J.; et al. Modulators of 14-3-3 protein-protein interactions. J. Med. Chem. 2018, 61, 3755–3778. [Google Scholar] [CrossRef] [Green Version]
- Litman, P.; Ohne, O.; Ben-Yaakov, S.; Shemesh-Darvish, L.; Yechezkel, T.; Salitra, Y.; Rubnov, S.; Cohen, I.; Senderowitz, H.; Kidron, D.; et al. A novel substrate mimetic inhibitor of PKB/Akt inhibits prostate cancer tumor growth in mice by blocking the PKB pathway. Biochemistry 2007, 46, 4716–4724. [Google Scholar] [CrossRef]
- Chassagnon, I.R.; McCarthy, C.A.; Chin, Y.K.; Pineda, S.S.; Keramidas, A.; Mobli, M.; Pham, V.; De Silva, T.M.; Lynch, J.W.; Widdop, R.E.; et al. Potent neuroprotection after stroke afforded by a double-knot spider-venom peptide that inhibits acid-sensing ion channel 1a. Proc. Natl. Acad. Sci. USA 2017, 114, 3750–3755. [Google Scholar] [CrossRef] [Green Version]
- Cafe-Mendes, C.C.; Ferro, E.S.; Britto, L.R.; Martins-de-Souza, D. Using mass spectrometry-based peptidomics to understand the brain and disorders such as Parkinson’s disease and schizophrenia. Curr. Top. Med. Chem. 2014, 14, 369–381. [Google Scholar] [CrossRef]
- Blackwell, H.E.; Grubbs, R.H. Highly efficient synthesis of covalently cross-linked peptide helices by ring-closing metathesis. Angew. Chem. Int. Ed. Engl. 1998, 37, 3281–3284. [Google Scholar] [CrossRef]
- Davies, J.S. The cyclization of peptides and depsipeptides. J. Pept. Sci. 2003, 9, 471–501. [Google Scholar] [CrossRef] [PubMed]
- Czabotar, P.E.; Lee, E.F.; van Delft, M.F.; Day, C.L.; Smith, B.J.; Huang, D.C.; Fairlie, W.D.; Hinds, M.G.; Colman, P.M. Structural insights into the degradation of Mcl-1 induced by BH3 domains. Proc. Natl. Acad. Sci. USA 2007, 104, 6217–6222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glas, A.; Bier, D.; Hahne, G.; Rademacher, C.; Ottmann, C.; Grossmann, T.N. Constrained peptides with target-adapted cross-links as inhibitors of a pathogenic protein-protein interaction. Angew. Chem. Int. Ed. Engl. 2014, 53, 2489–2493. [Google Scholar] [CrossRef]
- Assi, S.A.; Tanaka, T.; Rabbitts, T.H.; Fernandez-Fuentes, N. PCRPi: Presaging Critical Residues in Protein interfaces, a new computational tool to chart hot spots in protein interfaces. Nucl. Acids Res. 2010, 38, e86. [Google Scholar] [CrossRef] [Green Version]
- London, N.; Raveh, B.; Movshovitz-Attias, D.; Schueler-Furman, O. Can self-inhibitory peptides be derived from the interfaces of globular protein-protein interactions? Proteins 2010, 78, 3140–3149. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.B.; Howe, W.J. Computer design of bioactive molecules: A method for receptor-based de novo ligand design. Proteins 1991, 11, 314–328. [Google Scholar] [CrossRef] [PubMed]
- King, C.A.; Bradley, P. Structure-based prediction of protein-peptide specificity in Rosetta. Proteins 2010, 78, 3437–3449. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, D.; Clark, D.E.; Li, J.; Murray, C.W.; Robson, B.; Waszkowycz, B.; Westhead, D.R. PRO_LIGAND: An approach to de novo molecular design. 4. Application to the design of peptides. J. Comput.-Aided Mol. Des. 1995, 9, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Teixido, M.; Belda, I.; Rosello, X.; Gonzalez, S.; Fabre, M.; Llor, X.; Bacardit, J.; Garrell, J.M.; Vilaro, S.; Albericio, F.; et al. Development of a genetic algorithm to design and identify peptides that can cross the blood-brain barrier. QSAR Comb. Sci. 2003, 22, 745–753. [Google Scholar] [CrossRef]
- Diharce, J.; Cueto, M.; Beltramo, M.; Aucagne, V.; Bonnet, P. In silico peptide ligation: Iterative residue docking and linking as a new approach to predict protein-peptide interactions. Molecules 2019, 24, 1351. [Google Scholar] [CrossRef] [Green Version]
- Roberts, K.E.; Cushing, P.R.; Boisguerin, P.; Madden, D.R.; Donald, B.R. Computational design of a PDZ domain peptide inhibitor that rescues CFTR activity. PLoS Comput. Biol. 2012, 8, e1002477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidman, D.; Wolfson, H.J. PinaColada: Peptide-inhibitor ant colony ad-hoc design algorithm. Bioinformatics 2016, 32, 2289–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raveh, B.; London, N.; Schueler-Furman, O. Sub-angstrom modeling of complexes between flexible peptides and globular proteins. Proteins 2010, 78, 2029–2040. [Google Scholar] [CrossRef]
- Donsky, E.; Wolfson, H.J. PepCrawler: A fast RRT-based algorithm for high-resolution refinement and binding affinity estimation of peptide inhibitors. Bioinformatics 2011, 27, 2836–2842. [Google Scholar] [CrossRef] [Green Version]
- Watt, P.M. Screening for peptide drugs from the natural repertoire of biodiverse protein folds. Nat. Biotechnol. 2006, 24, 177–183. [Google Scholar] [CrossRef]
- Cruz-Migoni, A.; Fuentes-Fernandez, N.; Rabbitts, T.H. Peptides: Minimal drug surrogates to interrogate and interfere with protein function. MedChemComm 2013, 4, 1218–1221. [Google Scholar] [CrossRef]
- Badia-Villanueva, M.; Defaus, S.; Foj, R.; Andreu, D.; Oliva, B.; Sierra, A.; Fernandez-Fuentes, N. Evaluation of computationally designed peptides against TWEAK, a cytokine of the tumour necrosis factor ligand family. Int. J. Mol. Sci. 2021, 22, 1066. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkruys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Hurtado Del Pozo, C.; Prosper, F.; et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell 2020, 181, 905–913. [Google Scholar] [CrossRef]
- Chan, K.K.; Dorosky, D.; Sharma, P.; Abbasi, S.A.; Dye, J.M.; Kranz, D.M.; Herbert, A.S.; Procko, E. Engineering human ACE2 to optimize binding to the spike protein of SARS coronavirus 2. Science 2020, 369, 1261–1265. [Google Scholar] [CrossRef]
- Cao, L.; Goreshnik, I.; Coventry, B.; Case, J.B.; Miller, L.; Kozodoy, L.; Chen, R.E.; Carter, L.; Walls, A.C.; Park, Y.J.; et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science 2020, 370, 426–431. [Google Scholar] [CrossRef]
- Barnes, C.O.; Jette, C.A.; Abernathy, M.E.; Dam, K.A.; Esswein, S.R.; Gristick, H.B.; Malyutin, A.G.; Sharaf, N.G.; Huey-Tubman, K.E.; Lee, Y.E.; et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature 2020, 588, 682–687. [Google Scholar] [CrossRef]
- Shi, R.; Shan, C.; Duan, X.; Chen, Z.; Liu, P.; Song, J.; Song, T.; Bi, X.; Han, C.; Wu, L.; et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature 2020, 584, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Duyvesteyn, H.M.E.; Chen, C.P.; Huang, C.G.; Chen, T.H.; Shih, S.R.; Lin, Y.C.; Cheng, C.Y.; Cheng, S.H.; Huang, Y.C.; et al. Structural basis for the neutralization of SARS-CoV-2 by an antibody from a convalescent patient. Nat. Struct. Mol. Biol. 2020, 27, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Schoof, M.; Faust, B.; Saunders, R.A.; Sangwan, S.; Rezelj, V.; Hoppe, N.; Boone, M.; Billesbolle, C.B.; Puchades, C.; Azumaya, C.M.; et al. An ultrapotent synthetic nanobody neutralizes SARS-CoV-2 by stabilizing inactive Spike. Science 2020, 370, 1473–1479. [Google Scholar] [CrossRef]
- Song, Y.; Song, J.; Wei, X.; Huang, M.; Sun, M.; Zhu, L.; Lin, B.; Shen, H.; Zhu, Z.; Yang, C. Discovery of aptamers targeting the receptor-binding domain of the SARS-CoV-2 spike glycoprotein. Anal. Chem. 2020, 92, 9895–9900. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Y.L.; Wu, J.; Qi, J.; Zeng, Z.; Wan, Q.; Chen, Z.; Manandhar, P.; Cavener, V.S.; Boyle, N.R.; et al. Neutralizing aptamers block S/RBD-ACE2 interactions and prevent host cell Infection. Angew. Chem. Int. Ed. Engl. 2021, 60, 10273–10278. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Kral, P. Computational design of ACE2-based peptide inhibitors of SARS-CoV-2. ACS Nano 2020, 14, 5143–5147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Konduri, K.S.; Pattisapu, R.; Pattisapu, J.; Konduri, G.G.; Zwetchkenbaum, J.; Roy, B.; Barman, M.; Frazier, A.; Hurst, B.L.; Düzgüneş, N. ProLung budesonide inhibits SARS-CoV-2 replication and reduces lung inflammation. Arch. Pharmacol. Ther. 2021, 3, 52–65. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Peptide | Target | Experimental System | Reference |
|---|---|---|---|
| CBZ-D-Phe-L-Phe-Gly | gp41 fusion peptide | Cell–cell fusion; infection | [18] |
| gp41 N-terminal hexapeptide | gp41 fusion peptide | gp120/gp41 pseudo-virus fusion | [19] |
| aa 81–92 of CD4 | gp120 | Cell–cell fusion; infection | [20] |
| CD4 CDR-3-like domain | gp120 | Cell–cell fusion | [21] |
| V3 loop eight-branched peptide | gp120 | HIV infection | [22] |
| DP-178 | gp41 NHR | Cell–cell fusion | [23,24] |
| PEG-C34 | gp41 NHR | Cell–cell fusion | [25] |
| Cholesterol-C34 | gp41 NHR | Infection | [26] |
| PIE12-trimer | gp41 NHR hydrophobic pocket | HIV-1 and pseudo-type infection | [27] |
| Peptide | Target | Experimental System | Reference |
|---|---|---|---|
| SARS-CoV HR2 peptide | S-protein NHR (HR1) | Cytopathic effect | [39] |
| GST-HR2 peptide | S-protein NHR (HR1) | Cytopathic effect | [39] |
| HR1-1 | S-protein CHR (HR2) | Pseudo-typed HIV-luc/SARS virus | [40] |
| HR2-18 | S-protein NHR (HR1) | Pseudo-typed HIV-luc/SARS virus | [40] |
| CP-1 (from HR2) | S-protein HR1 domain | Infection | [37] |
| sHR2-8 | S-protein HR1 domain | Infection | [41] |
| P6 (from HR2) | Deep groove of HR1 trimer | S-expressing cell fusion with ACE2-cells | [42] |
| N46 (from HR1) | S-protein HR2 domain | S-expressing cell fusion with ACE2-cells | [42] |
| SR9 and SR9EK13 | S-protein HR1 domain | Entry at the plasma membrane | [43] |
| Peptide | Target | Experimental System | Reference |
|---|---|---|---|
| HR2P | S-protein NHR (HR1) | Virus replication; cell–cell fusion | [46] |
| HR2P-M2 | S-protein NHR (HR1) | Pseudo-virus infection; cell–cell fusion | [47] |
| HR2P-M2 | S-protein NHR (HR1) | Infection in a murine model | [47] |
| MERS-5HB | S-protein CHR (HR2) | Pseudo-virus infection; cell–cell fusion | [48] |
| EK1 | S-protein NHR (HR1) | Cell–cell fusion; infection | [49] |
| EK1C4 | S-protein NHR (HR1) | Cell–cell fusion; infection | [49] |
| SARS-CoV-2 HRC lipopeptide | S-protein NHR (HR1) | Cell–cell fusion; virus infection | [50] |
| Peptide | Target | Experimental System | Reference |
|---|---|---|---|
| EK1 | S-protein NHR (HR1) | Cell–cell fusion; pseudo-virus infection | [49] |
| EK1C4 | S-protein NHR (HR1) | Cell–cell fusion; pseudo-virus infection | [49] |
| IBP02 | S-protein NHR (HR1) | Cell–cell fusion; pseudo-virus infection | [51] |
| SARS-CoV-2 HRC lipopeptide | S-protein NHR (HR1) | Cell–cell fusion; virus infection | [50] |
| Peptide | Target | Experimental System | Reference |
|---|---|---|---|
| C18-s2 | HA receptor-binding domain | Virus infection | [61] |
| EB | HA receptor-binding domain | Virus-induced cytopathology | [62] |
| P155-181-chol | HA internal coiled coil | Virus infection | [64] |
| 43 aa Peptide-CPP | HA2 helix–helix interactions | Virus–liposome fusion; in vivo | [65] |
| iHA-100 | HA stalk domain | Virus infection | [67] |
| Cyclic P5, P6 | HA stem hydrophobic region | Virus neutralization | [68] |
| Peptide | Target | Experimental System | Reference |
|---|---|---|---|
| Myrcludex B (bulevirtide) | NTCP | Hepatitis B virus infection | [74,76] |
| E27 | E1–E2 protein dimerization | Hepatitis C virus fusion | [78] |
| CL58 | E2 | Hepatitis C virus fusion | [79] |
| Peptide | Target | Experimental System | Reference |
|---|---|---|---|
| DN57opt | Dengue virus E protein | Virus binding | [112] |
| 1OAN1 | Dengue virus E protein | Virus binding | [112] |
| P5 | JEV receptor | Virus infection; tissue pathology | [111] |
| Peptide | Target | Experimental System | Reference |
|---|---|---|---|
| It1b | HSV-1 membrane | Virus infection | [121] |
| MelN4 | HSV-1 membrane | Virus infection | [121] |
| gBh1m-Cys-PEG24-Chol | HSV-1 gB protein | Viral cytotoxicity | [115] |
| Peptide | Target | Experimental System | Reference |
|---|---|---|---|
| Tat-Ebo | EV GP2 protein | Pseudo-virus and virus infection | [127] |
| RVFV-6 | EV GP2 protein, EV membrane | Virus infection | [128] |
| Pep-3.2 | EV GPcl | EV-GP pseudo-virus | [129] |
| Pep-3.3 | EV GPcl | EV-GP pseudo-virus | [129] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Düzgüneş, N.; Fernandez-Fuentes, N.; Konopka, K. Inhibition of Viral Membrane Fusion by Peptides and Approaches to Peptide Design. Pathogens 2021, 10, 1599. https://doi.org/10.3390/pathogens10121599
Düzgüneş N, Fernandez-Fuentes N, Konopka K. Inhibition of Viral Membrane Fusion by Peptides and Approaches to Peptide Design. Pathogens. 2021; 10(12):1599. https://doi.org/10.3390/pathogens10121599
Chicago/Turabian StyleDüzgüneş, Nejat, Narcis Fernandez-Fuentes, and Krystyna Konopka. 2021. "Inhibition of Viral Membrane Fusion by Peptides and Approaches to Peptide Design" Pathogens 10, no. 12: 1599. https://doi.org/10.3390/pathogens10121599