
The Predictive Value of Mutation Screening for Anticipating COVID-19 Waves

 , , , , , , , , and
, , , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Selection
2.2. PCR Testing and Mutation Screening
2.3. WGS Methodology
2.4. Mathematical Modelling for VOC Prevalence Dynamics
3. Results
3.1. Validation of Mutation Screening Assays by Whole-Genome Sequencing
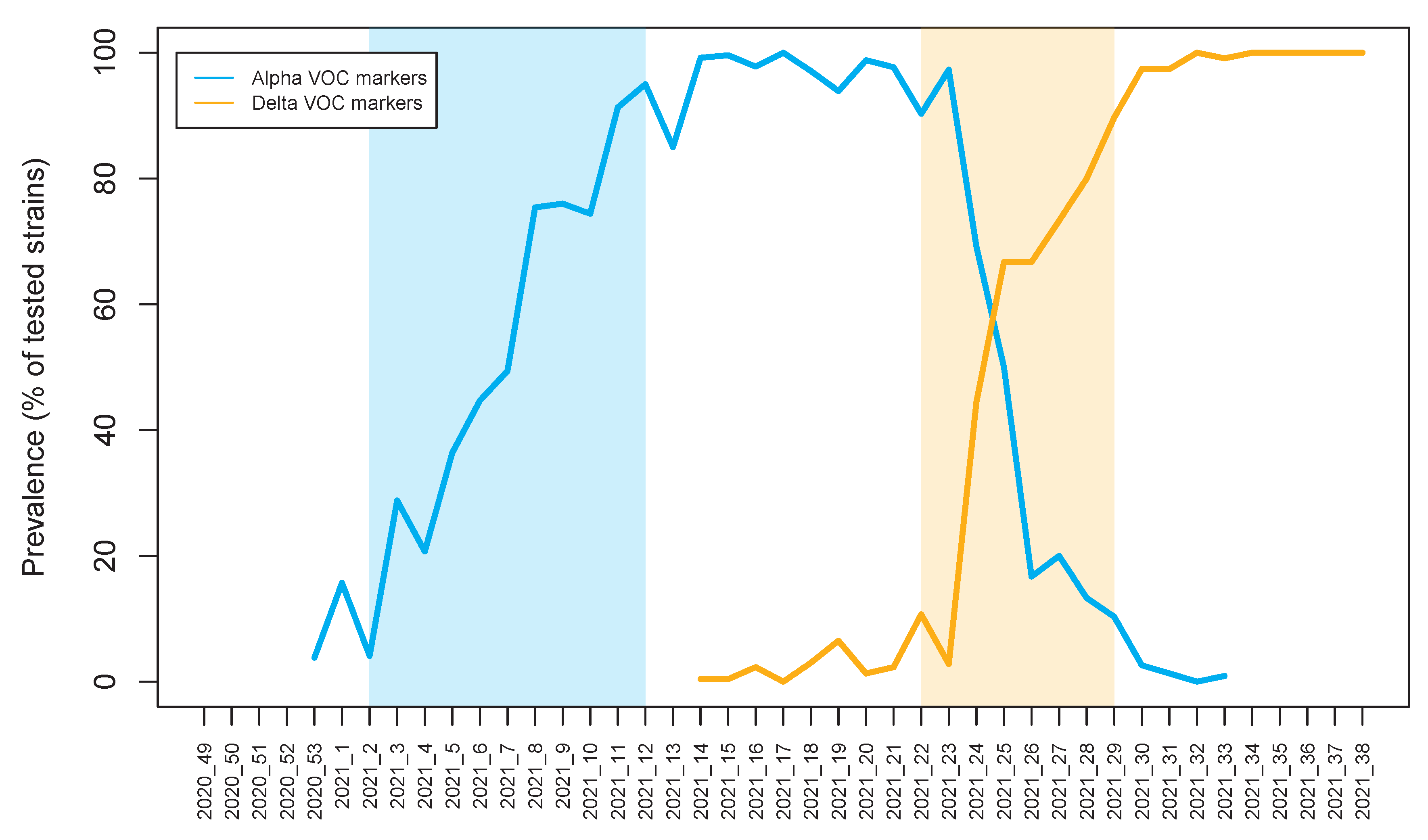
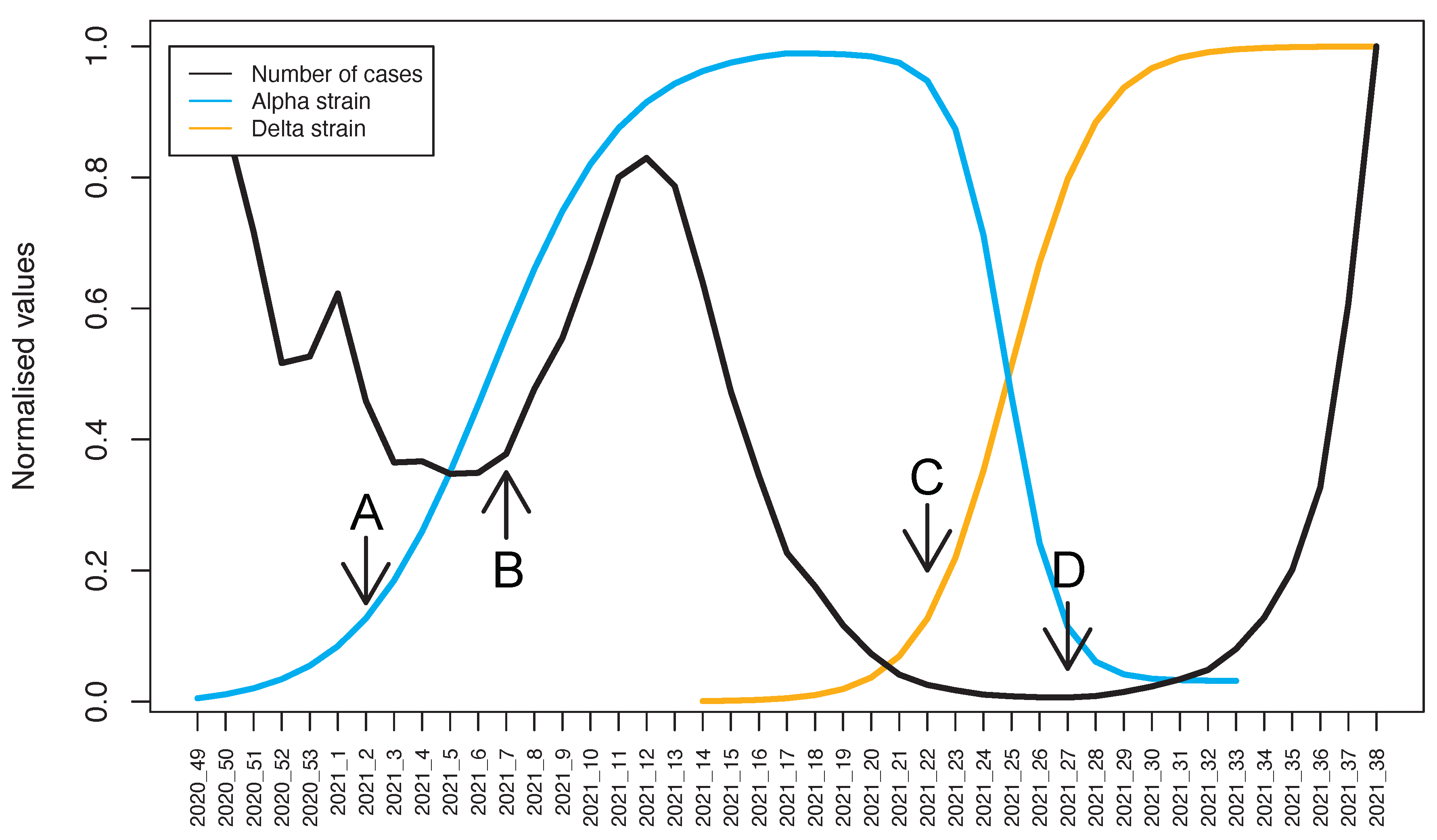
3.2. The Dynamics of Alpha and Delta VOC Prevalence
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| NGS | Next Generation Sequencing |
| SGTF | S-gene Target Failure |
| SNP | Single-nucleotide Polymorphism |
| VOC | Variant of Concern |
| WGS | Whole-Genome Sequencing |
References
- O’Toole, Á.; Hill, V.; Pybus, O.G.; Watts, A.; Bogoch, I.I.; Khan, K.; Messina, J.P.; Tegally, H.; Lessells, R.R.; Giandhari, J.; et al. Tracking the international spread of SARS-CoV-2 lineages B.1.1.7 and B.1.351/501Y-V2 [version 1; peer review: 3 approved] Network for Genomic Surveillance in South Africa ( NGS-SA ), Danish Covid-19 Genome Consortium ( DCGC ), Communicable. Wellcome Open Res. 2021, 26, 6. [Google Scholar]
- Cov-lineages.org. Global Report-B.1.617.2. 2021. Available online: https://cov-lineages.org/global_report_B.1.617.2.html (accessed on 1 November 2021).
- Public Health England. SARS-CoV-2 Variants of Concern and Variants under Investigation in England. Technical Report June. 2021. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/993879/Variants_of_Concern_VOC_Technical_Briefing_15.pdf (accessed on 1 November 2021).
- Boehm, E.; Kronig, I.; Neher, R.A.; Eckerle, I.; Vetter, P.; Kaiser, L.; Geneva Centre for Emerging Viral Diseases. Novel SARS-CoV-2 variants: The pandemics within the pandemic. Clin. Microbiol. Infect. 2021, 27, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- CNSCBT. COVID-19 Case Analysis. 2021. Available online: https://www.cnscbt.ro/index.php/analiza-cazuri-confirmate-covid19/2730-raport-saptamanal-episaptamana39-2021/file (accessed on 1 November 2021).
- Alizon, S.; Haim-Boukobza, S.; Foulongne, V.; Verdurme, L.; Trombert-Paolantoni, S.; Lecorche, E.; Roquebert, B.; Sofonea, M.T. Rapid spread of the SARS-CoV-2 Delta variant in some French regions, June 2021. Eurosurveillance 2021, 26, 2100573. [Google Scholar] [CrossRef] [PubMed]
- ECDC. Methods for the Detection and Identification of SARS-CoV-2 Variants Summary Diagnostic Screening Assays of Known VOCs S-Gene Drop out or Target Failure. Technical Report. 2021. Available online: https://www.ecdc.europa.eu/sites/default/files/documents/Methods-for-the-detection-and-identification-of-SARS-CoV-2-variants-WHO-ECDC.pdf (accessed on 1 November 2021).
- Surleac, M.; Casangiu, C.; Banica, L.; Milu, P.; Florea, D.; Sandulescu, O.; Streinu-Cercel, A.; Vlaicu, O.; Tudor, A.; Hohan, R.; et al. Short Communication:Evidence of Novel SARS-CoV-2 Variants Circulation in Romania. AIDS Res. Hum. Retroviruses 2021, 37, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Baranyi, J.; Roberts, T. A dynamic approach to predicting bacterial growth in food. Int. J. Food Microbiol. 1994, 23, 277–294. [Google Scholar] [CrossRef]
- Sofonea, M.T.; Reyné, B.; Elie, B.; Djidjou-Demasse, R.; Selinger, C.; Michalakis, Y.; Alizon, S. Memory is key in capturing COVID-19 epidemiological dynamics. Epidemics 2021, 35, 100459. [Google Scholar] [CrossRef] [PubMed]
- Li, P.E.; Lo, C.C.; Anderson, J.J.; Davenport, K.W.; Bishop-Lilly, K.A.; Xu, Y.; Ahmed, S.; Feng, S.; Mokashi, V.P.; Chain, P.S.G. Enabling the democratization of the genomics revolution with a fully integrated web-based bioinformatics platform. Nucleic Acids Res. 2017, 45, 67–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhulst, P.F. Notice sur la loi que la population suit dans son accroissement. Quetelet 1838, 10, 113–121. [Google Scholar]
- Elzhov, T.V.; Mullen, K.M.; Spiess, A.N.; Bolker, B. Package ‘minpack.lm’-R Interface to the Levenberg-Marquardt Nonlinear Least-Squares Algorithm Found in MINPACK, Plus Support for Bounds. 2015. Available online: https://cran.r-project.org/web/packages/minpack.lm/index.html (accessed on 1 November 2021).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372. [Google Scholar] [CrossRef]
- Gonzalez-Parra, G.; Martínez-Rodríguez, D.; Villanueva-Micó, R.J. Impact of a New SARS-CoV-2 Variant on the Population: A Mathematical Modeling Approach. Math. Comput. Appl. 2021, 26, 25. [Google Scholar] [CrossRef]
- Shah, S.A.; Moore, E.; Robertson, C.; McMenamin, J.; Katikireddi, S.V.; Simpson, C.R.; Shi, T.; Agrawal, U.; McCowan, C.; Stock, S.; et al. Predicted COVID-19 positive cases, hospitalisations, and deaths associated with the Delta variant of concern, June–July, 2021. Lancet. Digit. Health 2021, 3, e539–e541. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Test for Alpha VOC | Target | Sensitivity | Specificity | PPV | NPV | F1-Score |
|---|---|---|---|---|---|---|
| TaqPath (n = 120) | SGTF (del 69/70) | 89% | 97% | 98% | 85% | 0.93 |
| TaqMan SNP (n = 166) | 501Y | 100% | 92% | 95% | 100% | 0.97 |
| Test for Delta VOC | Target | Sensitivity | Specificity | PPV | NPV | F1-Score |
|---|---|---|---|---|---|---|
| TaqMan SNP (n = 166) | 501N | 100% | 95% | 89% | 100% | 0.94 |
| Allplex Variants II (n = 25) | 452R | 100% | 100% | 100% | 100% | 1 |
| TaqMan SNP (n = 38) | 681R | 100% | 100% | 100% | 100% | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hohan, R.; Milu, P.; Paraschiv, S.; Casangiu, C.; Tudor, A.; Vlaicu, O.; Banica, L.; Surleac, M.; Florea, D.; Otelea, D. The Predictive Value of Mutation Screening for Anticipating COVID-19 Waves. Pathogens 2021, 10, 1464. https://doi.org/10.3390/pathogens10111464
Hohan R, Milu P, Paraschiv S, Casangiu C, Tudor A, Vlaicu O, Banica L, Surleac M, Florea D, Otelea D. The Predictive Value of Mutation Screening for Anticipating COVID-19 Waves. Pathogens. 2021; 10(11):1464. https://doi.org/10.3390/pathogens10111464
Chicago/Turabian StyleHohan, Robert, Petre Milu, Simona Paraschiv, Corina Casangiu, Andreea Tudor, Ovidiu Vlaicu, Leontina Banica, Marius Surleac, Dragos Florea, and Dan Otelea. 2021. "The Predictive Value of Mutation Screening for Anticipating COVID-19 Waves" Pathogens 10, no. 11: 1464. https://doi.org/10.3390/pathogens10111464