2.2. Reversion Categories
For the reverse transcriptase and protease regions of the pol gene, the patients were divided based on the degree to which they returned to the wild type. These categories are high reversion, low reversion, and non-reversion. We established the reversion group assignment on the return to wild-type pattern in the reverse transcriptase region of the plasma sequences. The decision rule for assignment to each group was based on a comparison of the number of mutations that the patient experienced between baseline (week 0) and week 12. The “None” group had an equal or greater number of mutations at the end than at the baseline. The “Low” group had fewer but not zero mutations. In other words, they had at least one mutation but less than they had at baseline. The “High” group experienced zero mutations at week 12 after showing one or more at baseline. In this scheme, the controls were those with zero mutations at baseline.
The last line of
Table 1 shows the count of patients classified in each reversion group, and
Figure 1 shows the average number of mutations per patient for each of the groups within the four classes of blood fraction (PBMC or plasma) and region on the pol gene (reverse transcriptase or protease). The “All Resistant Subjects” column includes all three reversion groups.
2.3. Viral Load Dynamics
Table 1 also shows the evolution of viral load (measured by a log
10 transformation of the raw data) over the twelve weeks of the study for all four groups (three reversion groups and the control group). The data underlying these results, the viral loads, CD4+ counts, and CD8+ counts for all patients at all sample collection times are included in the
Supplementary Materials, Table S2. The means and standard deviations by groups are provided in
Table S2B.
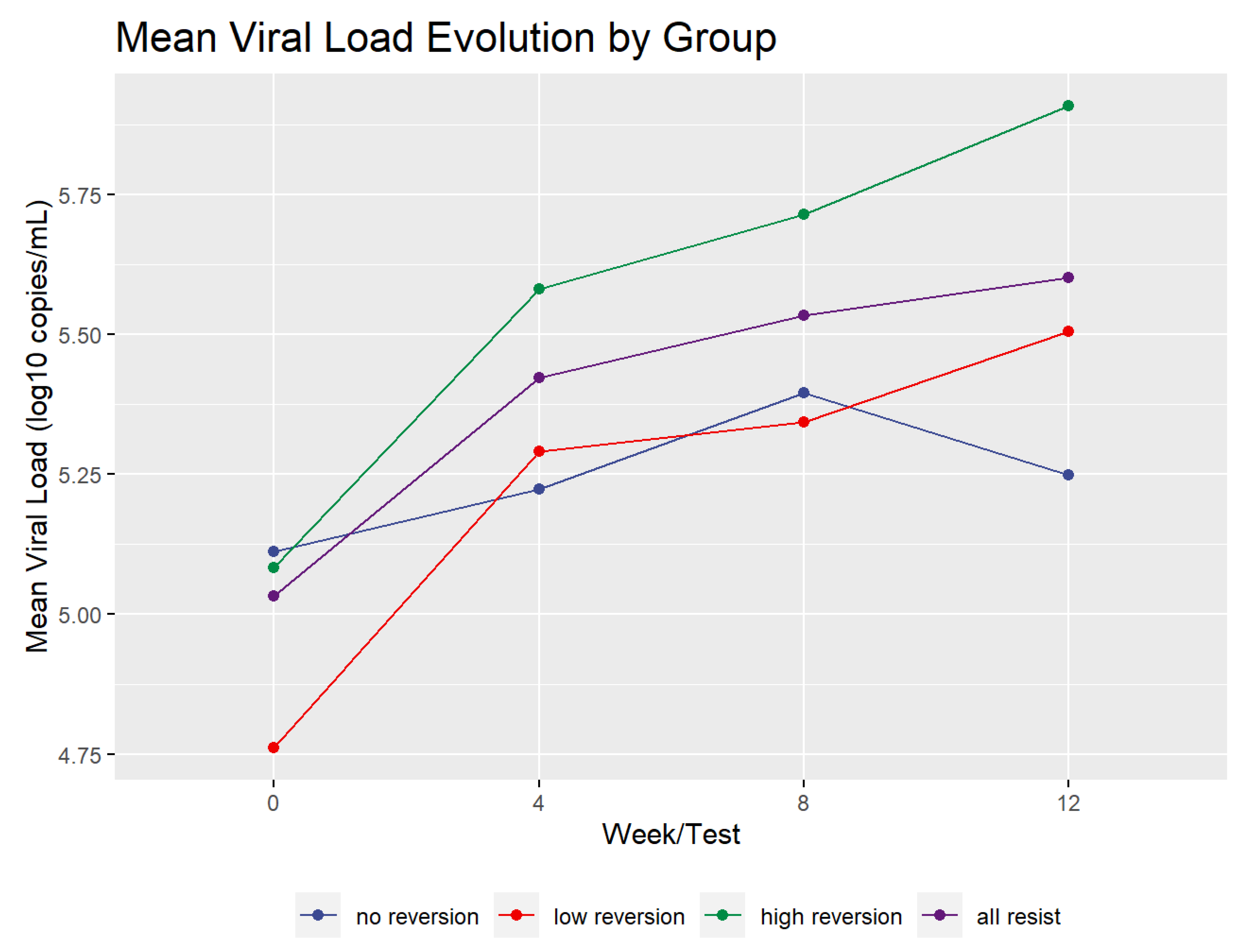
Figure 2 shows the mean profile of the viral load dynamics for all of the reversion groups. The mean viral load increased during the test period for the high and low groups by more than 0.5 log
10. However, the viral load of the no reversion group rose until week 8 and then declined. It only increased over the study period by 0.136 log
10. Overall, the groups, including the controls, increased by an average of 0.568 log
10. In addition, the high and low reversion groups’ viral load increase was statistically significant. A paired two-sample t-test of the difference between viral load in all resistant patients (groups High, Low, and No taken together) at baseline and at week 12 had a value of t = 6.2919 with 35 degrees of freedom and
p < 1.0 × 10
−6. The linear increase in viral load in the current cohort contrasts with the rapid rise in viral load reported in a previous study [
9].
2.4. CD4+ and CD8+ T Cell Dynamics
Figure 3 shows the effect of treatment interruption on CD4+ T cell counts of patients in the three reversion and control groups. In this case, patients showed an immediate and consistent decline in cell counts over the 12 weeks of the experiment. The decrease was an average of 99.0 cells/mL among patients showing drug-resistant mutations at baseline. By the end of week 12, the three reversion groups showed mean CD4+ levels below 200 cells/mL.
Focusing on the three groups that showed initial resistance mutations, the decline in CD4 levels between baseline and week 12 was statically significant (Wilcoxon signed-rank test with continuity correction, p-value < 1.0 × 10−6.
The CD8+ T cell results roughly mirrored those of CD4+ cells for the low reversion and control groups. The no reversion and high reversion groups showed slight overall declines, with the no reversion group showing a slight increase between weeks 4 and 8. Overall, the resistant groups experienced a mean decrease in CD8+ levels of 145.4 cells per mL with a standard deviation of 409.7 cells. The
Supplementary Materials contain the figure showing the CD8+ results (
Figure S1).
2.5. Reversion to Wild Type among Resistant Patients
The number of mutations declined during the ATI, as expected for the reversion groups in both plasma and PBMC blood fractions and in both the reverse transcriptase and protease genomic regions. However, only in reverse transcriptase mutations in plasma (high reversion group) did the number of mutations go to zero. In all other cases, there remained a small number of mutations per patient in week 12.
That the resistant mutations would revert to the wild-type virus due to ATI among individuals with antiretroviral failure was only partially supported by the data from this study where the proviral DNA profile is concerned. We limited all of the analyses of reversion to wild type to the 36 patients who showed resistant mutations. However, we include in the
Supplementary Materials, Table S3, the mutations pre- and post-ATI for all of the patients as well as the antiretroviral drugs (including the different regimens) that they used before the ATI. This table also specifies the subtypes of HIV for each patient.
2.6. Reversion to Wild Type in Plasma
The plasma sequences collected at baseline and study end (week 12) show an uneven reversion pattern despite the expectation that all mutated codons would revert to wild type after 12 weeks of ATI. Among the patients showing drug resistance, a mean of 14.95 codons evolved between the start and end of the treatment interruption (95% confidence interval of 10.61 to 19.28). Of the 434 amino acids in the sequence studied, 111 different codons evolved during the study. Some of these 111 codons had variant amino acids at the end of the survey, so the number of different mutations during the treatment interruption totaled 164. For example, codon 20 showed an evolution of R10K in five patients and I10K in two other patients.
We focused our analysis on those codons that evolved in more than one patient.
Table 2 lists these codons. We have not included codon changes that came from only one patient, as they may be as much a result of a sampling or measurement anomaly as they are of an underlying biological process. The format for referring to the amino acids shows the mutated state (pre-ATI) first and then the evolved state, which is the wild-type amino acid for that location in all cases. For example, the listing for codon at position pr10 is generally referred to as L10I, with the wild-type amino acid coming before the location number and the mutation after. The “pr” designation refers to protease inhibitor resistant codons and “tr” refers to reverse transcriptase resistant codons.
Although our sample was drawn well before IAS began to report drug resistance mutations, we tested whether mutations in patients with drug resistance are consistent with the 2017 IAS list of drug-resistant mutations. In 35 of the 40 codons in the table (87.5%), the patients reverted to wild type as defined in the IAS 2017 interpretations.
2.7. Protease Inhibitor (PI) Mutations at DNA Level
We conducted a parallel analysis of the PBMC samples from the 36 resistant patients for the evolution of both protease inhibitor and reverse transcriptase inhibitor resistance mutations. We hypothesized that the high reversion level that we would see at the plasma level would not be observed in this compartment.
Of the PI codons on the IAS list, we initially had data on 22. These were codons 10, 20, 24, 30, 32, 36, 46, 47, 48, 50, 53, 54, 63, 71, 73, 77, 82, 84, 85, 88, 89, 90, and 93. We eliminated codons 32, 47, and 50 from our analysis as they either had 0 or 1 tests that showed that mutation and therefore did not allow comparison. For codon 89, only patient 3 exhibited a mutation. Codons 77 and 88 showed an increase in the number of patients exhibiting the mutation during the 12 weeks and thus had positive slopes in the regression. Accordingly, it was not possible to calculate a time to 0 patients using regression. However, the Kaplan–Meier survival analysis result showed a probability of the survival of the mutation at codon 77 of 0.318 (95% CI of 0.173–0.587) and at codon 88 of 0.176 (95% CI of 0.063–0.493). Given the overall sample size of 36 and the variable number of patients who showed the mutation (between 7 and 11 for codon 77, and between 1 and 4 for codon 88), it is not possible to tell if this result is due to a sampling error or to some fundamental observation about these codons.
We evaluated the consistency of results between the time to 0 mutations for each codon and the survival analysis. We found a correlation between these values of 0.69. This correlation was significant (
p = 0.00013) using a Pearson’s Product Moment Correlation test. We included a figure with this result in the
Supplementary Materials (Figure S2).
Table 3 shows the number of weeks it would take for each mutated codon to return to wild type at the DNA level via a regression line projection for all three types of drug resistance. Rather than being a specific forecast of how long the mutations would persist in the absence of ART, the projection indicates the mutation’s general tendency. The strength of the regression model for each codon is expressed in the R
2 column. This measure indicates the percentage of overall variance that the model expresses. The table also shows the probability that the mutation would survive after the 12-week interruption derived from the Kaplan–Meier analysis. In other words, it estimates the likelihood that the mutation would not revert to wild type. As seen in the table, those codons with smaller mutation survival probabilities also had generally shorter projected periods to 0 patients carrying the mutation.
The mean number of weeks for the protease inhibitor mutations is 34.5 weeks with a standard deviation of 27.3 weeks.
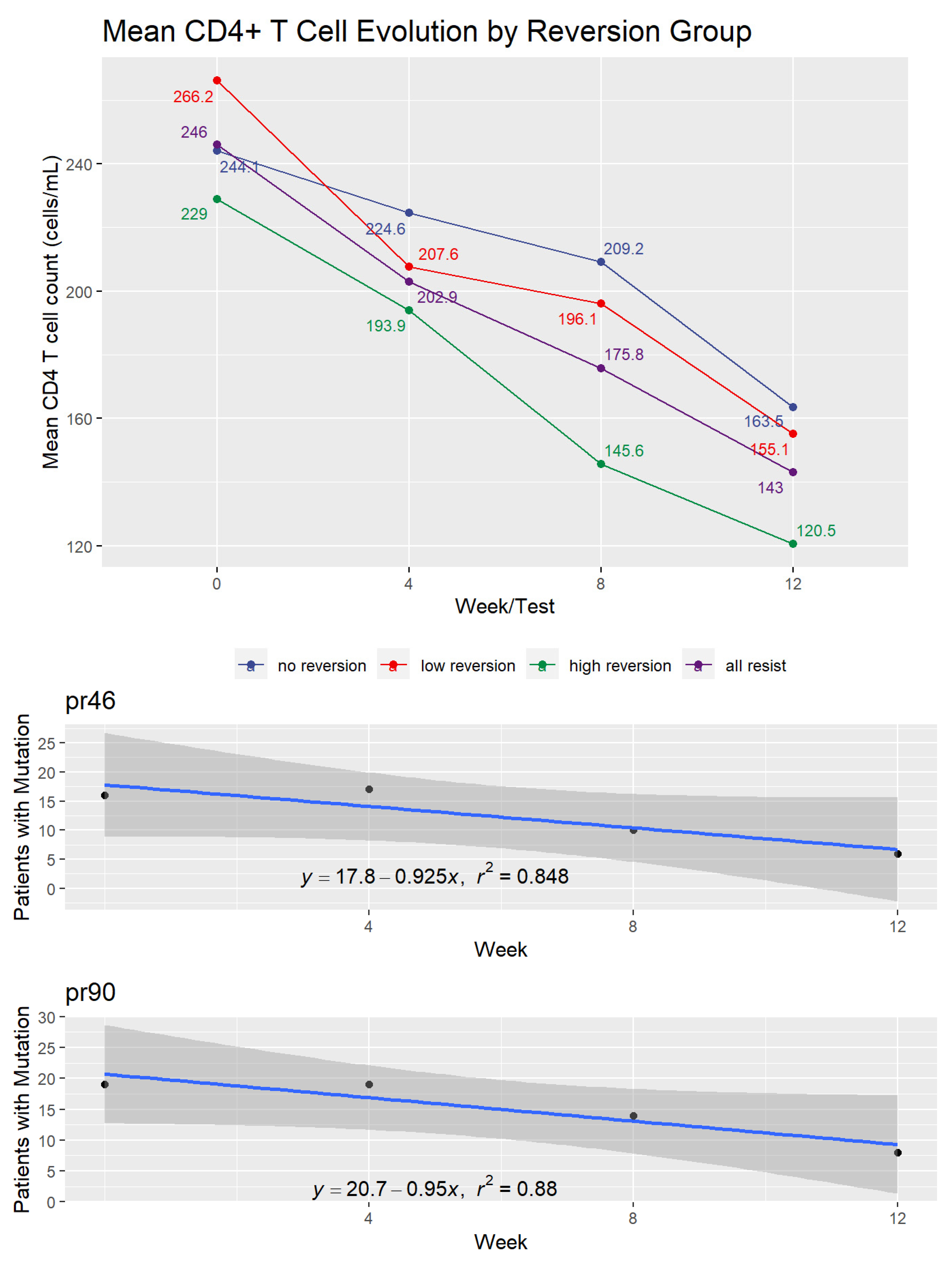
Figure 4 illustrates the transition to wild type virus for codons 46 and 90. These were chosen for display because they have the highest coefficients of determination (R
2) among the PI mutation codons. Graphs for the other codons are found in the data repository for the study (
https://github.com/jameshunterbr/ATI, accessed on 5 October 2021).
This figure shows the number of patients with a mutation at each test and a regression of these points. The regression equation shows the parameters determining the line, and the R2 indicates the degree to which the line explains the points.
2.8. Nucleoside Reverse Transcriptase Inhibitor (NRTI) Mutations at DNA Level
We had data on nine of the IAS’s list of 15 NRTI codons (41, 65, 67, 70, 75, 184, 210, 215, and 219). We eliminated codon 65 from further analysis as it had only one patient with a mutation at this location at week 0. Codon 75 just had two data points: one in week 0 and one at week 12.
Table 4 shows the trend toward reversion to wild type for the NRTI codons for the remaining seven codons and the probability that the mutation would survive after the 12-week ATI. The overall mean for reversion time was 20.9 weeks with a standard deviation of 4.8 weeks.
Codon 184 (M184V) comes first in the Stanford University database of NRTI mutations because of its 100-fold reduction in patient susceptibility to Lamivudine/Emtricitabine but its enhancement of the susceptibility to Zidovudine and Tenofovir Disoproxil Fumarate [
37]. The other six NRTI mutations in our study are all thymidine analog mutations (TAMs).
Figure 5 shows the number of weeks that we project it would take to completely extinguish the M184V mutation and the two groups of TAM mutations (TAM 1 including the mutations at codons 41, 210, and 215 and TAM 2 including codons 67, 70, and 219). The R
2 measure indicates the relative variability within the group.
Both TAM groups have mean values very close to each other. According to a sample of 3 per group, there is no difference between the two TAM types with a Wilcoxon Rank Sum test (
p = 1.0). However, the TAMs considered together take a significantly longer time to revert to wild type than codon 184 according to a one-sided Wilcoxon Signed Rank test (
p = 0.016). This started with 28 patients exhibiting the Valine mutation, the loss of one carbon atom that distinguishes it from the Isoleucine wild type. By the end of the 12 weeks, the number showing this mutation had dropped to 8, and our linear regression model for this codon projected it would be eliminated by week 16. The TAM groups showed fewer patients with the mutations at the start of the interruption (20.7 for TAM 1 and 17.7 for TAM 2), declining to 8.3 and 7.7, respectively, at the end of the study. The higher rate of reversion for I184V than the TAMS can be seen in greater detail in the panels of
Figure 5, which show all of the NRTI codons.
2.9. Non-Nucleoside Reverse Transcriptase Inhibitor (NNRTI) Mutations at DNA Level
Our data included 7 of the 16 non-nucleoside reverse transcriptase inhibitors (NNRTI) on the IAS list (103, 106, 108, 179, 181, 188, and 190). We eliminated codons 106 and 108 from the analysis as they had insufficient data to permit a regression to be performed. We also removed codons 179 and 188 from the analysis as the counts of patients with the mutation only varied between 1 and 3, which strongly suggested that our sample was not typical of the population. The last line of
Table 5 shows the number of weeks required for the three remaining NNRTI mutations to revert to wild type.
The average number of weeks for NNRTI mutations to revert to wild type was 19.8 weeks with a standard deviation of 2.2 weeks.
Figure 6 shows the rate of reversion for the NNRTI mutations.
Although the graphs and tables we display here show the range of rates of reversion to wild type among the codons studied, the average for each of the three classes of antiretroviral medications indicates that this is a prolonged process that may last as long as 8 months (in the case of protease inhibitor mutations).
Table 4 shows these averages.
2.10. CD38 and HLA-DR T Cell Activation Markers
The study sample showed a greater presence of CD38+ and HLA-DR+ proteins on CD8+ T cells in patients who returned to wild type. The no reversion group, whose mutations remained constant despite the treatment interruption, expressed a significantly lower level of these cell activation biomarkers (measured by the percentage of CD8+ T-cells with both CD38 and HLA-DR present) than did the high reversion group.
Table 5 and
Figure 7 show these summary measures.
This result parallels the observation that cell activation markers would be expressed in greater quantity in untreated HIV-1 patients than in patients receiving treatment [
32].
To create an index of cell activity absent viral load, we corrected the cell activation marker levels for the log
10 viral load by dividing the cell activation markers’ expression by the log
10 viral load. As
Table 5 and the second panel of
Figure 7 show, there is still a difference among the groups, but it is small and not statistically significant.
Table 6 uses the mean of all four visits to determine the cell activation level. If we look at the evolution of the cell activation from baseline to week 12 in the no reversion and high reversion groups (where the difference was significant above), we find that there is an apparent difference between the two groups, but it is not significant when calculated for either the raw values or the corrected cell activation values, as seen in
Table 6. While the No Reversion group shows more significant growth in cell activation between baseline and the ATI end, the difference is not significant.
We compared the T cell activation level with the viral load and CD4+ T-cell levels for all the patients. Overall, the correlation with viral load was low but statistically significant (Spearman’s rank correlation, r = 0.46,
p-value < 1.0 × 10
−6). However, the correlation between cell activation and CD4+ T-cell levels is stronger and statistically significant (Spearman’s rank correlation, r = −0.52,
p-value < 1.0 × 10
−6) showing that the higher the T cell activation, the lower the CD4+ T-cell counts.
Figure 8 shows these correlations.
Previous research has found a positive association between viral load and CD38+ CD8+ T cells [
31]. Our correlation results do not show the same level of significance in such an association.
However, our data regarding cell activation and CD4+ T cell counts closely parallel previous studies analyzing cell activation in subpopulations of HIV patients [
31,
38]. Our data mirror those of treatment-naive patients in these studies. In the current study, patients at baseline are under treatment, and at the end of the study, they should be similar to untreated patients in these other studies.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}