
Variable Proportions of Phylogenetic Clustering and Low Levels of Antiviral Drug Resistance among the Major HBV Sub-Genotypes in the Middle East and North Africa


 ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
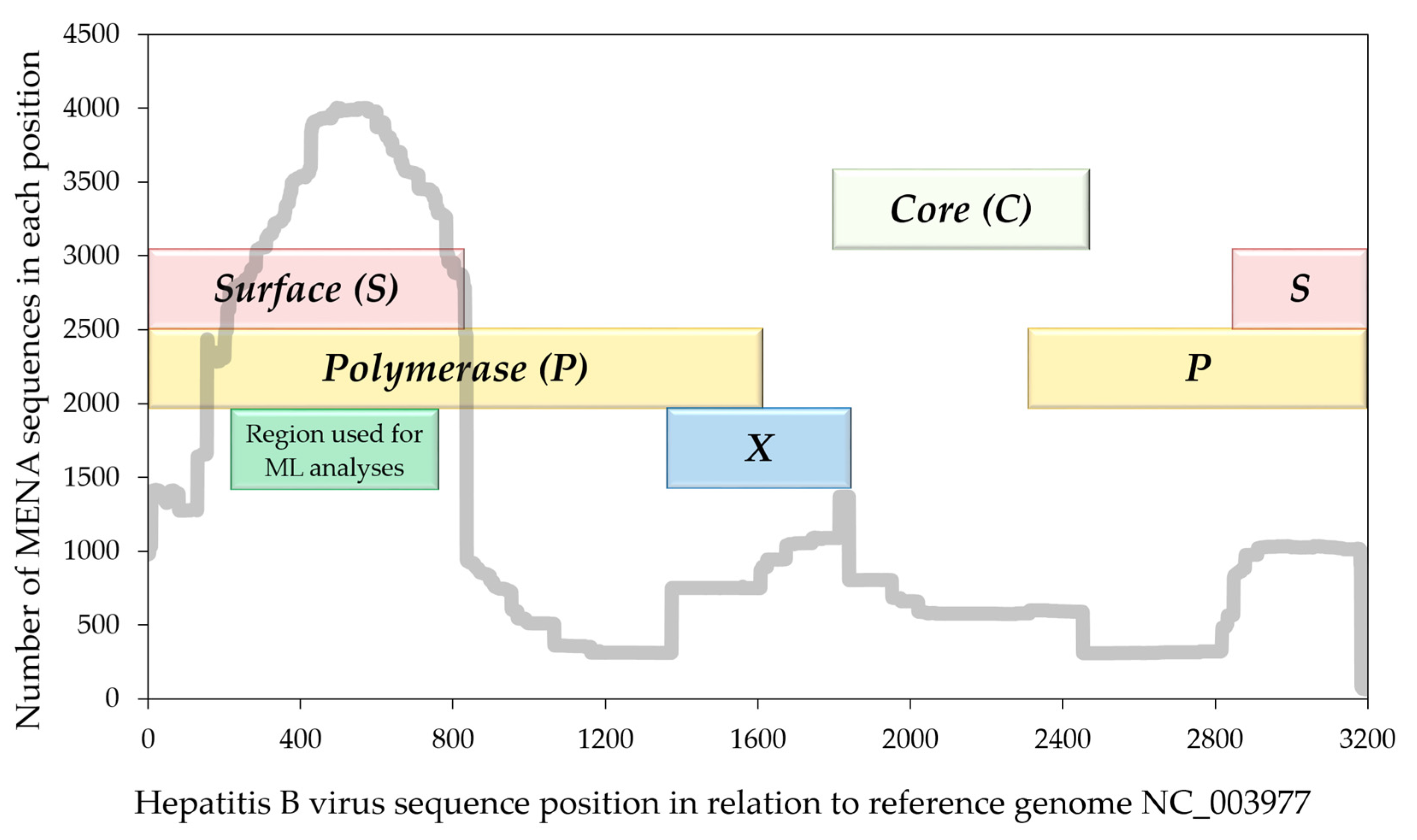
2.1. Study Design and Sequence Inclusion Criteria
2.2. Determination of HBV Genotype Distribution, Antiviral Resistance and S Gene Mutations
2.3. Analysis of HBV Domestic Transmission in the MENA Region
2.4. Statistical Analysis
3. Results
3.1. Characteristics of the MENA HBV Molecular Dataset
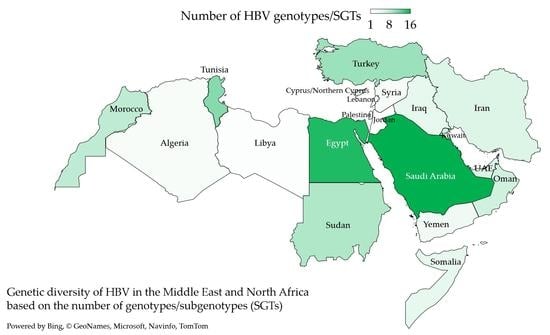
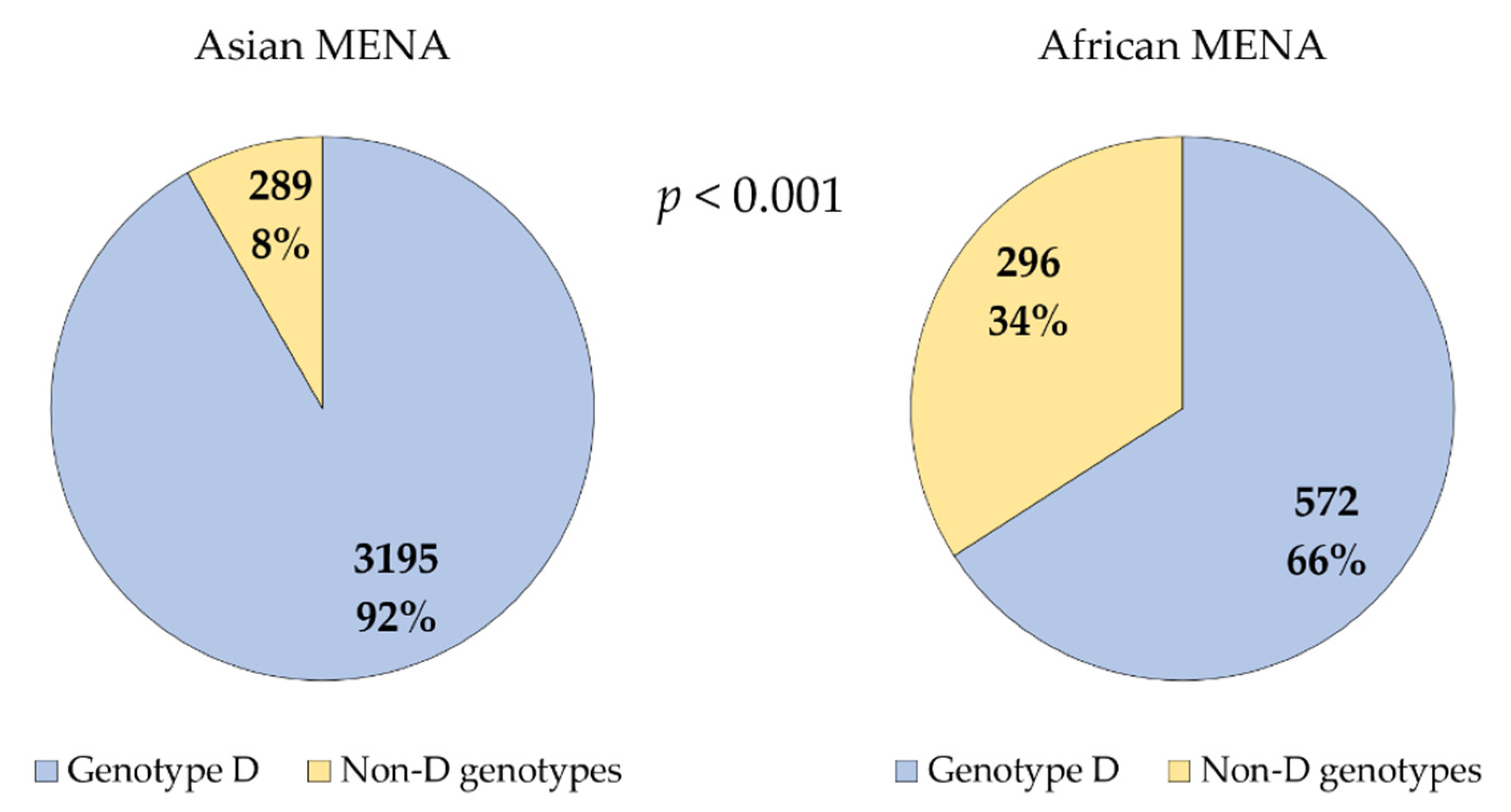
3.2. Genotype D Dominated HBV Infections in the MENA despite the Detection of an Extensive Genetic Diversity
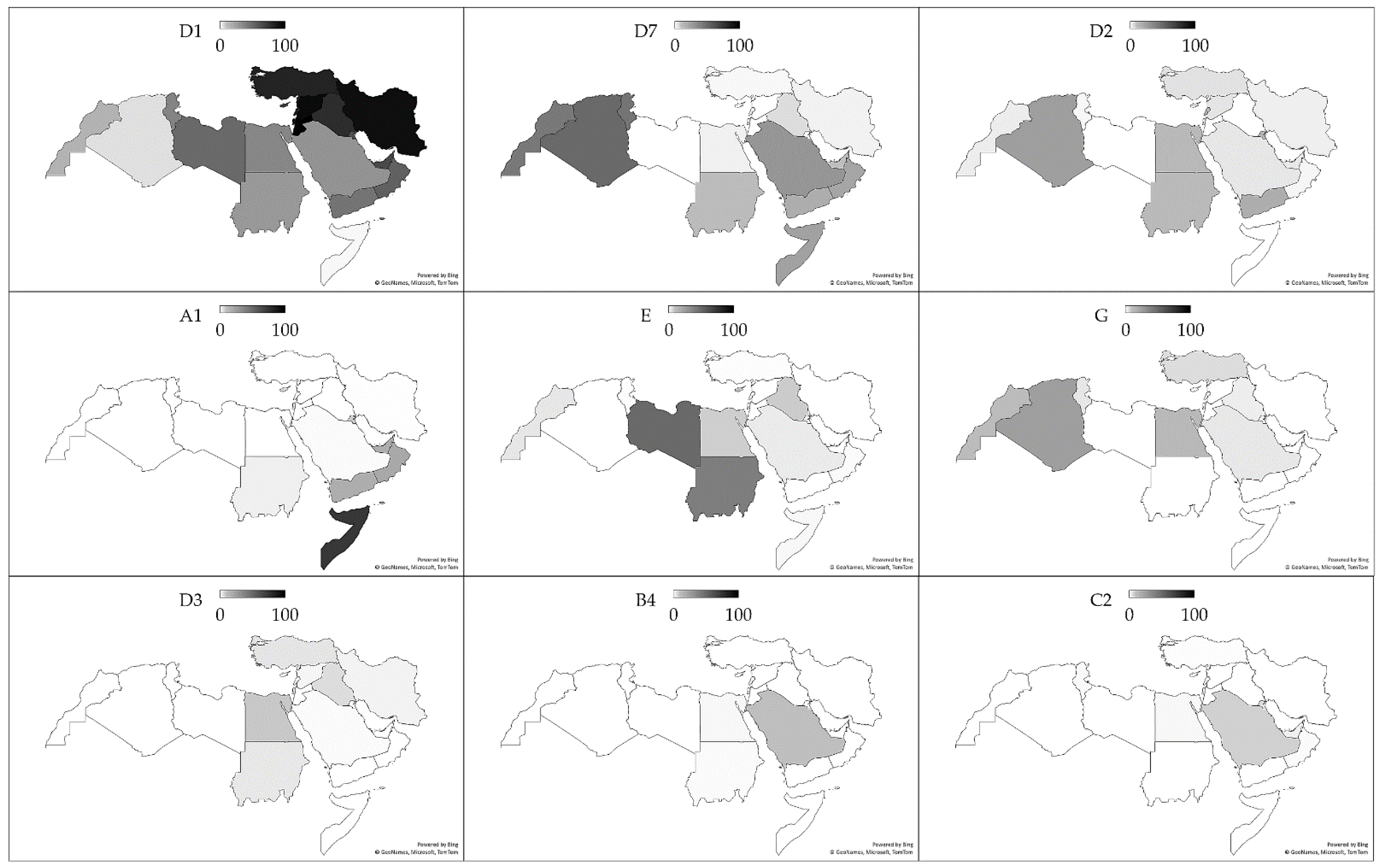
3.3. HBV Sub-Genotype Distribution in the MENA Region
3.4. Antiviral Drug Resistance and S Gene Mutations in the MENA HBV Sequences
3.5. Variable Proportions of Putative Domestic HBV Transmission in the MENA Region for Different SGTs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- The World Health Organization (WHO). Hepatitis B—Key Facts. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-b (accessed on 14 June 2021).
- Nelson, N.P.; Easterbrook, P.J.; McMahon, B.J. Epidemiology of Hepatitis B Virus Infection and Impact of Vaccination on Disease. Clin. Liver Dis. 2016, 20, 607–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Lancet Global Health Editorial. The hidden threat of hepatitis B. Lancet Glob. Health 2016, 4, 502. [Google Scholar] [CrossRef] [Green Version]
- Schillie, S.; Vellozzi, C.; Reingold, A.; Harris, A.; Haber, P.; Ward, J.W.; Nelson, N.P. Prevention of Hepatitis B Virus Infection in the United States: Recommendations of the Advisory Committee on Immunization Practices. MMWR Recomm. Rep. 2018, 67, 1–31. [Google Scholar] [CrossRef]
- Schinazi, R.F.; Ehteshami, M.; Bassit, L.; Asselah, T. Towards HBV curative therapies. Liver Int. 2018, 38, 102–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revill, P.; Testoni, B.; Locarnini, S.; Zoulim, F. Global strategies are required to cure and eliminate HBV infection. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, R.; Kottilil, S. Strategies to eliminate HBV infection. Future Virol. 2014, 9, 565–585. [Google Scholar] [CrossRef] [Green Version]
- Sayan, M.; Akhan, S.C. Antiviral drug-associated potential vaccine-escape hepatitis B virus mutants in Turkish patients with chronic hepatitis B. Int. J. Infect. Dis. 2011, 15, 722–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayagam, S.; Thursz, M.; Sicuri, E.; Conteh, L.; Wiktor, S.; Low-Beer, D.; Hallett, T.B. Requirements for global elimination of hepatitis B: A modelling study. Lancet Infect. Dis. 2016, 16, 1399–1408. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization (WHO). Global Health Sector Strategy on Viral Hepatitis 2016–2021. Towards Ending Viral Hepatitis. Available online: https://apps.who.int/iris/bitstream/handle/10665/246177/WHO?sequence=1 (accessed on 20 June 2021).
- Chotiyaputta, W.; Lok, A.S. Hepatitis B virus variants. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 453–462. [Google Scholar] [CrossRef]
- Kidd-Ljunggren, K.; Miyakawa, Y.; Kidd, A.H. Genetic variability in hepatitis B viruses. J. Gen. Virol. 2002, 83, 1267–1280. [Google Scholar] [CrossRef]
- Kay, A.; Zoulim, F. Hepatitis B virus genetic variability and evolution. Virus Res. 2007, 127, 164–176. [Google Scholar] [CrossRef]
- Zhou, Y.; Holmes, E.C. Bayesian estimates of the evolutionary rate and age of hepatitis B virus. J. Mol. Evol. 2007, 65, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Sunbul, M. Hepatitis B virus genotypes: Global distribution and clinical importance. World J. Gastroenterol. 2014, 20, 5427–5434. [Google Scholar] [CrossRef]
- Pavesi, A. Origin, Evolution and Stability of Overlapping Genes in Viruses: A Systematic Review. Genes 2021, 12, 809. [Google Scholar] [CrossRef]
- Li, S.; Wang, Z.; Li, Y.; Ding, G. Adaptive evolution of proteins in hepatitis B virus during divergence of genotypes. Sci. Rep. 2017, 7, 1990. [Google Scholar] [CrossRef]
- Lin, Y.-Y.; Liu, C.; Chien, W.-H.; Wu, L.-L.; Tao, Y.; Wu, D.; Lu, X.; Hsieh, C.-H.; Chen, P.-J.; Wang, H.-Y.; et al. New insights into the evolutionary rate of hepatitis B virus at different biological scales. J. Virol. 2015, 89, 3512–3522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pourkarim, M.R.; Amini-Bavil-Olyaee, S.; Kurbanov, F.; Van Ranst, M.; Tacke, F. Molecular identification of hepatitis B virus genotypes/subgenotypes: Revised classification hurdles and updated resolutions. World J. Gastroenterol. 2014, 20, 7152–7168. [Google Scholar] [CrossRef] [PubMed]
- Kurbanov, F.; Tanaka, Y.; Mizokami, M. Geographical and genetic diversity of the human hepatitis B virus. Hepatol. Res. 2010, 40, 14–30. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.T.; Trinh, T.N.; Abe, K. New complex recombinant genotype of hepatitis B virus identified in Vietnam. J. Virol. 2008, 82, 5657–5663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatematsu, K.; Tanaka, Y.; Kurbanov, F.; Sugauchi, F.; Mano, S.; Maeshiro, T.; Nakayoshi, T.; Wakuta, M.; Miyakawa, Y.; Mizokami, M. A genetic variant of hepatitis B virus divergent from known human and ape genotypes isolated from a Japanese patient and provisionally assigned to new genotype J. J. Virol. 2009, 83, 10538–10547. [Google Scholar] [CrossRef] [Green Version]
- MacLachlan, J.H.; Cowie, B.C. Hepatitis B virus epidemiology. Cold Spring Harb. Perspect. Med. 2015, 5, 021410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, J.J.; Stevens, G.A.; Groeger, J.; Wiersma, S.T. Global epidemiology of hepatitis B virus infection: New estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine 2012, 30, 2212–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polaris Observatory Collaborators. Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: A modelling study. Lancet Gastroenterol. Hepatol. 2018, 3, 383–403. [Google Scholar] [CrossRef]
- Velkov, S.; Ott, J.J.; Protzer, U.; Michler, T. The Global Hepatitis B Virus Genotype Distribution Approximated from Available Genotyping Data. Genes 2018, 9, 495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zampino, R.; Boemio, A.; Sagnelli, C.; Alessio, L.; Adinolfi, L.E.; Sagnelli, E.; Coppola, N. Hepatitis B virus burden in developing countries. World J Gastroenterol. 2015, 21, 11941–11953. [Google Scholar] [CrossRef]
- Schaefer, S. Hepatitis B virus: Significance of genotypes. J. Viral Hepat. 2005, 12, 111–124. [Google Scholar] [CrossRef]
- Tanwar, S.; Dusheiko, G. Is there any value to hepatitis B virus genotype analysis? Curr. Gastroenterol. Rep. 2012, 14, 37–46. [Google Scholar] [CrossRef]
- Thakur, V.; Guptan, R.C.; Kazim, S.N.; Malhotra, V.; Sarin, S.K. Profile, spectrum and significance of HBV genotypes in chronic liver disease patients in the Indian subcontinent. J. Gastroenterol. Hepatol. 2002, 17, 165–170. [Google Scholar] [CrossRef]
- Mayerat, C.; Mantegani, A.; Frei, P.C. Does hepatitis B virus (HBV) genotype influence the clinical outcome of HBV infection? J. Viral Hepat. 1999, 6, 299–304. [Google Scholar] [CrossRef]
- Halfon, P.; Bourlière, M.; Pol, S.; Benhamou, Y.; Ouzan, D.; Rotily, M.; Khiri, H.; Renou, C.; Pénaranda, G.; Saadoun, D.; et al. Multicentre study of hepatitis B virus genotypes in France: Correlation with liver fibrosis and hepatitis B e antigen status. J. Viral Hepat. 2006, 13, 329–335. [Google Scholar] [CrossRef]
- Heiberg, I.L.; Hoegh, M.; Ladelund, S.; Niesters, H.G.; Hogh, B. Hepatitis B virus DNA in saliva from children with chronic hepatitis B infection: Implications for saliva as a potential mode of horizontal transmission. Pediatr. Infect. Dis. J. 2010, 29, 465–467. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, H.; Inui, A.; Fujisawa, T. The role of body fluids in the horizontal transmission of hepatitis B virus via household/close contact. EMJ 2016, 1, 68–75. [Google Scholar]
- Atkins, M.; Nolan, M. Sexual transmission of hepatitis B. Curr. Opin. Infect. Dis. 2005, 18, 67–72. [Google Scholar] [CrossRef]
- McKee, M.; Keulertz, M.; Habibi, N.; Mulligan, M.; Woertz, E. Demographic and economic material factors in the MENA region. Middle East and North Africa Regional Architecture: Mapping Geopolitical Shifts, Regional Order and Domestic Transformations. Work. Pap. 2017, 3, 43. [Google Scholar]
- Sallam, M.; Sahin, G.O.; Ingman, M.; Widell, A.; Esbjornsson, J.; Medstrand, P. Genetic characterization of human immunodeficiency virus type 1 transmission in the Middle East and North Africa. Heliyon 2017, 3, 00352. [Google Scholar] [CrossRef] [Green Version]
- Sallam, M.; Ababneh, N.A.; Dababseh, D.; Bakri, F.G.; Mahafzah, A. Temporal increase in D614G mutation of SARS-CoV-2 in the Middle East and North Africa. Heliyon 2021, 7, 06035. [Google Scholar] [CrossRef]
- Fallahian, F.; Najafi, A. Epidemiology of hepatitis C in the Middle East. Saudi J. Kidney Dis. Transpl. 2011, 22, 1–9. [Google Scholar]
- Chaabna, K.; Cheema, S.; Abraham, A.; Alrouh, H.; Lowenfels, A.B.; Maisonneuve, P.; Mamtani, R. Systematic overview of hepatitis C infection in the Middle East and North Africa. World J. Gastroenterol. 2018, 24, 3038–3054. [Google Scholar] [CrossRef] [PubMed]
- Al-Sadeq, D.W.; Taleb, S.A.; Zaied, R.E.; Fahad, S.M.; Smatti, M.K.; Rizeq, B.R.; Al Thani, A.A.; Yassine, H.M.; Nasrallah, G.K. Hepatitis B Virus Molecular Epidemiology, Host-Virus Interaction, Coinfection, and Laboratory Diagnosis in the MENA Region: An Update. Pathogens 2019, 8, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamoud, Y.A.; Miller, F.D.; Abu-Raddad, L.J. Potential for human immunodeficiency virus parenteral transmission in the Middle East and North Africa: An analysis using hepatitis C virus as a proxy biomarker. World J. Gastroenterol. 2014, 20, 12734–12752. [Google Scholar] [CrossRef]
- Nojoomi, F.; Ghasemian, A.; Fallahi, S.; Hasanvand, F. High prevalence and risk factors of hepatitis B, C and E infections among Middle Eastern countries. Immunopathol. Persa 2018, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Sharafi, H.; Alavian, S.M. The Rising Threat of Hepatocellular Carcinoma in the Middle East and North Africa Region: Results from Global Burden of Disease Study 2017. Clin. Liver Dis. 2019, 14, 219–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madihi, S.; Syed, H.; Lazar, F.; Zyad, A.; Benani, A. A Systematic Review of the Current Hepatitis B Viral Infection and Hepatocellular Carcinoma Situation in Mediterranean Countries. Biomed. Res. Int. 2020, 2020, 7027169. [Google Scholar] [CrossRef]
- El Chaar, M.; El Jisr, T.; Allain, J.P. Hepatitis B virus DNA splicing in Lebanese blood donors and genotype A to E strains: Implications for hepatitis B virus DNA quantification and infectivity. J. Clin. Microbiol. 2012, 50, 3159–3167. [Google Scholar] [CrossRef] [Green Version]
- Khaled, I.A.; Mahmoud, O.M.; Saleh, A.F.; Bioumie, E.E. Prevalence of HBV genotypes among Egyptian hepatitis patients. Mol. Biol. Rep. 2011, 38, 4353–4357. [Google Scholar] [CrossRef]
- Salem, M.A.; Elnifro, E.M.; Alshuwen, F. Molecular analysis of hepatitis B virus isolates in Libya: Predominant circulation of hepatitis B virus genotype D. J. Gastroenterol. Hepatol. Res. 2012, 1, 119–121. [Google Scholar]
- Gourari, S.; Brichler, S.; Le Gal, F.; Abdou-Chekaraou, M.; Beloufa, M.A.; Khelifa, R.; Djaballah, H.; Boufekane, M.; Nani, A.; Afredj, N.; et al. Hepatitis B virus and hepatitis delta virus subtypes circulating in Algeria and seroprevalence of HDV infection. J. Med. Virol. 2019, 91, 72–80. [Google Scholar] [CrossRef] [Green Version]
- Ababneh, N.A.; Sallam, M.; Kaddomi, D.; Attili, A.M.; Bsisu, I.; Khamees, N.; Khatib, A.; Mahafzah, A. Patterns of hepatitis B virus S gene escape mutants and reverse transcriptase mutations among genotype D isolates in Jordan. PeerJ 2019, 7, 6583. [Google Scholar] [CrossRef] [Green Version]
- Nodeh, M.M.; Mosavat, A.; Valizadeh, N.; Zadeh, A.M.; Boskabadi, A.; Mashkani, B.; Sima, H.; Rafatpanah, H. Genotype characteristic and phylogenetic analysis of hepatitis B virus in northeast-Iran. Infect. Genet. Evol. 2018, 59, 148–154. [Google Scholar] [CrossRef]
- Khodadad, N.; Seyedian, S.S.; Moattari, A.; Biparva Haghighi, S.; Pirmoradi, R.; Abbasi, S.; Makvandi, M. In silico functional and structural characterization of hepatitis B virus PreS/S-gene in Iranian patients infected with chronic hepatitis B virus genotype D. Heliyon 2020, 6, 04332. [Google Scholar] [CrossRef]
- Sumer, U.; Sayan, M. Molecular Epidemiology of Hepatitis B Virus in Turkish Cypriot. Pol. J. Microbiol. 2019, 68, 449–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paraskevis, D.; Magiorkinis, G.; Magiorkinis, E.; Ho, S.Y.; Belshaw, R.; Allain, J.P.; Hatzakis, A. Dating the origin and dispersal of hepatitis B virus infection in humans and primates. Hepatology 2013, 57, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Kostaki, E.G.; Karamitros, T.; Stefanou, G.; Mamais, I.; Angelis, K.; Hatzakis, A.; Kramvis, A.; Paraskevis, D. Unravelling the history of hepatitis B virus genotypes A and D infection using a full-genome phylogenetic and phylogeographic approach. eLife 2018, 7, 36709. [Google Scholar] [CrossRef] [PubMed]
- Yousif, M.; Kramvis, A. Genotype D of hepatitis B virus and its subgenotypes: An update. Hepatol. Res. 2013, 43, 355–364. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Global Progress Report on HIV, Viral Hepatitis and Sexually Transmitted Infections. Available online: https://www.who.int/publications/i/item/9789240027077 (accessed on 10 October 2021).
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2016, 44, D67–D72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann-Fraune, M.; Beggel, B.; Kaiser, R.; Obermeier, M. Hepatitis B Virus Drug Resistance Tools: One Sequence, Two Predictions. Intervirology 2014, 57, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- McGinnis, S.; Madden, T.L. BLAST: At the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004, 32, W20–W25. [Google Scholar] [CrossRef]
- Anisimova, M.; Gascuel, O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst. Biol. 2006, 55, 539–552. [Google Scholar] [CrossRef]
- Anisimova, M.; Gil, M.; Dufayard, J.-F.; Dessimoz, C.; Gascuel, O. Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes. Syst. Biol. 2011, 60, 685–699. [Google Scholar] [CrossRef]
- Matsuo, J.; Do, S.H.; Yamamoto, C.; Nagashima, S.; Chuon, C.; Katayama, K.; Takahashi, K.; Tanaka, J. Clustering infection of hepatitis B virus genotype B4 among residents in Vietnam, and its genomic characters both intra- and extra-family. PLoS ONE 2017, 12, 0177248. [Google Scholar] [CrossRef] [Green Version]
- Hassan, A.S.; Pybus, O.G.; Sanders, E.J.; Albert, J.; Esbjörnsson, J. Defining HIV-1 transmission clusters based on sequence data. AIDS 2017, 31, 1211–1222. [Google Scholar] [CrossRef] [PubMed]
- Ragonnet-Cronin, M.; Hodcroft, E.; Hué, S.; Fearnhill, E.; Delpech, V.; Brown, A.J.L.; Lycett, S. Automated analysis of phylogenetic clusters. BMC Bioinform. 2013, 14, 317. [Google Scholar] [CrossRef] [Green Version]
- Esbjörnsson, J.; Mild, M.; Audelin, A.; Fonager, J.; Skar, H.; Bruun Jørgensen, L.; Liitsola, K.; Björkman, P.; Bratt, G.; Gisslén, M.; et al. HIV-1 transmission between MSM and heterosexuals, and increasing proportions of circulating recombinant forms in the Nordic Countries. Virus Evol. 2016, 2, vew010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sallam, M.; Esbjörnsson, J.; Baldvinsdóttir, G.; Indriðason, H.; Björnsdóttir, T.B.; Widell, A.; Gottfreðsson, M.; Löve, A.; Medstrand, P. Molecular epidemiology of HIV-1 in Iceland: Early introductions, transmission dynamics and recent outbreaks among injection drug users. Infect. Genet. Evol. 2017, 49, 157–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization (WHO). Technical Paper the Growing Threats of Hepatitis B and C in the Eastern Mediterranean Region: A Call for Action. Available online: https://applications.emro.who.int/docs/EM_RC56_3_en.pdf (accessed on 9 September 2021).
- Pourkarim, M.R.; Razavi, H.; Lemey, P.; Van Ranst, M. Iran’s hepatitis elimination programme is under threat. Lancet 2018, 392, 1009. [Google Scholar] [CrossRef] [Green Version]
- Specialist Panel on Chronic Hepatitis B in the Middle East. A review of chronic hepatitis B epidemiology and management issues in selected countries in the Middle East. J. Viral Hepat. 2012, 19, 9–22. [Google Scholar] [CrossRef]
- Pybus, O.G.; Rambaut, A. Evolutionary analysis of the dynamics of viral infectious disease. Nat. Rev. Genet. 2009, 10, 540–550. [Google Scholar] [CrossRef]
- Paraskevis, D.; Beloukas, A.; Stasinos, K.; Pantazis, N.; de Mendoza, C.; Bannert, N.; Meyer, L.; Zangerle, R.; Gill, J.; Prins, M.; et al. HIV-1 molecular transmission clusters in nine European countries and Canada: Association with demographic and clinical factors. BMC Med. 2019, 17, 4. [Google Scholar] [CrossRef] [Green Version]
- Alkaiyat, A.; Weiss, M.G. HIV in the Middle East and North Africa: Priority, culture, and control. Int. J. Public Health 2013, 58, 927–937. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.L.; Kao, J.H. The clinical implications of hepatitis B virus genotype: Recent advances. J. Gastroenterol. Hepatol. 2011, 26, 123–130. [Google Scholar] [CrossRef]
- Rajoriya, N.; Combet, C.; Zoulim, F.; Janssen, H.L.A. How viral genetic variants and genotypes influence disease and treatment outcome of chronic hepatitis B. Time for an individualised approach? J. Hepatol. 2017, 67, 1281–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jazayeri, S.M.; Carman, W.F. Evolution of Hepatitis B Genotype D in the Middle East and South Asia. Hepat. Mon. 2009, 9, 9–11. Available online: https://www.sid.ir/en/journal/ViewPaper.aspx?id=140258 (accessed on 20 June 2021).
- Norder, H.; Courouce, A.M.; Coursaget, P.; Echevarria, J.M.; Lee, S.D.; Mushahwar, I.K.; Robertson, B.H.; Locarnini, S.; Magnius, L.O. Genetic diversity of hepatitis B virus strains derived worldwide: Genotypes, subgenotypes, and HBsAg subtypes. Intervirology 2004, 47, 289–309. [Google Scholar] [CrossRef] [PubMed]
- Haghshenas, M.R.; Arabi, M.; Mousavi, T. Hepatitis B genotypes in iran. Mater. Socio-Med. 2014, 26, 129–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelnabi, Z.; Saleh, N.; Baraghithi, S.; Glebe, D.; Azzeh, M. Subgenotypes and Mutations in the S and Polymerase Genes of Hepatitis B Virus Carriers in the West Bank, Palestine. PLoS ONE 2014, 9, 113821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayan, M.; Dogan, C. Genotype/subgenotype distribution of hepatitis B virus among hemodialysis patients with chronical hepatitis B. Ann. Hepatol. 2012, 11, 849–854. [Google Scholar] [CrossRef]
- Aghakhani, A.; Hamkar, R.; Zamani, N.; Eslamifar, A.; Banifazl, M.; Saadat, A.; Sofian, M.; Adibi, L.; Irani, N.; Mehryar, M.; et al. Hepatitis B virus genotype in Iranian patients with hepatocellular carcinoma. Int. J. Infect. Dis. 2009, 13, 685–689. [Google Scholar] [CrossRef] [Green Version]
- Pourkarim, M.R.; Vergote, V.; Amini-Bavil-Olyaee, S.; Sharifi, Z.; Sijmons, S.; Lemey, P.; Maes, P.; Alavian, S.M.; Van Ranst, M. Molecular characterization of hepatitis B virus (HBV) strains circulating in the northern coast of the Persian Gulf and its comparison with worldwide distribution of HBV subgenotype D1. J. Med. Virol. 2014, 86, 745–757. [Google Scholar] [CrossRef]
- Khraif, R.M.; Salam, A.A.; Nair, P.S.; Elsegaey, I. Migration in Saudi Arabia: Present and Prospects. In India’s Low-Skilled Migration Middle East: Policies, Politics and Challenges; Palgrave Macmillan: Singapore, 2019; pp. 99–123. [Google Scholar] [CrossRef]
- Al-Qudari, A.Y.; Amer, H.M.; Abdo, A.A.; Hussain, Z.; Al-Hamoudi, W.; Alswat, K.; Almajhdi, F.N. Surface gene variants of hepatitis B Virus in Saudi Patients. Saudi J. Gastroenterol. 2016, 22, 133–138. [Google Scholar] [CrossRef]
- Zohry, A. The place of Egypt in the regional migration system as a receiving country. Rev. Eur. Des. Migr. Int. 2003, 19, 129–149. [Google Scholar] [CrossRef]
- Saudy, N.; Sugauchi, F.; Tanaka, Y.; Suzuki, S.; Aal, A.A.; Zaid, M.A.; Agha, S.; Mizokami, M. Genotypes and phylogenetic characterization of hepatitis B and delta viruses in Egypt. J. Med. Virol. 2003, 70, 529–536. [Google Scholar] [CrossRef]
- Arıkan, A.; Şanlıdağ, T.; Süer, K.; Sayan, M.; Akçalı, S.; Güler, E. Molecular epidemiology of hepatitis B virus in Northern Cyprus. Mikrobiyol. Bul. 2016, 50, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Al-Qahtani, A.A.; Pourkarim, M.R.; Trovão, N.S.; Vergote, V.; Li, G.; Thijssen, M.; Abdo, A.A.; Sanai, F.M.; Dela Cruz, D.; Bohol, M.F.F.; et al. Molecular epidemiology, phylogenetic analysis and genotype distribution of hepatitis B virus in Saudi Arabia: Predominance of genotype D1. Infect. Genet. Evol. 2020, 77, 104051. [Google Scholar] [CrossRef]
- Zhang, Z.-H.; Wu, C.-C.; Chen, X.-W.; Li, X.; Li, J.; Lu, M.-J. Genetic variation of hepatitis B virus and its significance for pathogenesis. World J. Gastroenterol. 2016, 22, 126–144. [Google Scholar] [CrossRef] [PubMed]
- Ciccozzi, M.; Chaouch, H.; Lo Presti, A.; Taffon, S.; Villano, U.; Equestre, M.; Bruni, R.; Marcantonio, C.; Tritarelli, E.; Cella, E.; et al. Evolutionary dynamics of HBV-D7 subgenotype in Tunisia. J. Med. Virol. 2017, 89, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Ezzikouri, S.; Pineau, P.; Benjelloun, S. Hepatitis B virus in the Maghreb Region: From epidemiology to prospective research. Liver Int. 2013, 33, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Rebbani, K.; Ouladlahsen, A.; Bensghir, A.; Akil, A.; Lamdini, H.; Issouf, H.; Brahim, I.; Kitab, B.; Fakhir, F.Z.; Wakrim, L.; et al. Co-infections with hepatitis B and C viruses in human immunodeficiency virus-infected patients in Morocco. Clin. Microbiol. Infect. 2013, 19, E454–E457. [Google Scholar] [CrossRef] [Green Version]
- Meldal, B.H.M.; Moula, N.M.; Barnes, I.H.A.; Boukef, K.; Allain, J.-P. A novel hepatitis B virus subgenotype, D7, in Tunisian blood donors. J. Gen. Virol. 2009, 90, 1622–1628. [Google Scholar] [CrossRef] [PubMed]
- Baha, W.; Ennaji, M.M.; Lazar, F.; Melloul, M.; El Fahime, E.; El Malki, A.; Bennani, A. HBV genotypes prevalence, precore and basal core mutants in Morocco. Infect. Genet. Evol. 2012, 12, 1157–1162. [Google Scholar] [CrossRef]
- Croagh, C.M.; Desmond, P.V.; Bell, S.J. Genotypes and viral variants in chronic hepatitis B: A review of epidemiology and clinical relevance. World J. Hepatol. 2015, 7, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Wai, C.T.; Fontana, R.J.; Polson, J.; Hussain, M.; Shakil, A.O.; Han, S.H.; Davern, T.J.; Lee, W.M.; Lok, A.S. Clinical outcome and virological characteristics of hepatitis B-related acute liver failure in the United States. J. Viral Hepat. 2005, 12, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Mina, T.; Amini Bavil Olyaee, S.; Tacke, F.; Maes, P.; Van Ranst, M.; Pourkarim, M.R. Genomic Diversity of Hepatitis B Virus Infection Associated with Fulminant Hepatitis B Development. Hepat. Mon. 2015, 15, 29477. [Google Scholar] [CrossRef] [Green Version]
- Zehender, G.; Ebranati, E.; Gabanelli, E.; Shkjezi, R.; Lai, A.; Sorrentino, C.; Lo Presti, A.; Basho, M.; Bruno, R.; Tanzi, E.; et al. Spatial and temporal dynamics of hepatitis B virus D genotype in Europe and the Mediterranean Basin. PLoS ONE 2012, 7, 37198. [Google Scholar] [CrossRef] [Green Version]
- Ciccozzi, M.; Ciccaglione, A.R.; Lo Presti, A.; Equestre, M.; Cella, E.; Ebranati, E.; Gabanelli, E.; Villano, U.; Bruni, R.; Yalcinkaya, T.; et al. Evolutionary dynamics of HBV-D1 genotype epidemic in Turkey. J. Med. Virol. 2014, 86, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Spitz, N.; Mello, F.C.A.; Moreira, A.S.; Gusatti, C.S.; Martins, R.M.B.; Gomes, S.A.; Bello, G.; Araujo, N.M. Reconstruction of the spatial and temporal dynamics of hepatitis B virus genotype D in the Americas. PLoS ONE 2019, 14, 0220342. [Google Scholar] [CrossRef]
- Yousif, M.; Mudawi, H.; Hussein, W.; Mukhtar, M.; Nemeri, O.; Glebe, D.; Kramvis, A. Genotyping and virological characteristics of hepatitis B virus in HIV-infected individuals in Sudan. Int. J. Infect. Dis 2014, 29, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Ingasia, L.A.O.; Kostaki, E.G.; Paraskevis, D.; Kramvis, A. Global and regional dispersal patterns of hepatitis B virus genotype E from and in Africa: A full-genome molecular analysis. PLoS ONE 2020, 15, 0240375. [Google Scholar] [CrossRef]
- Garmiri, P.; Rezvan, H.; Abolghasemi, H.; Allain, J.P. Full genome characterization of hepatitis B virus strains from blood donors in Iran. J. Med. Virol. 2011, 83, 948–952. [Google Scholar] [CrossRef]
- Shepard, C.W.; Simard, E.P.; Finelli, L.; Fiore, A.E.; Bell, B.P. Hepatitis B virus infection: Epidemiology and vaccination. Epidemiol. Rev. 2006, 28, 112–125. [Google Scholar] [CrossRef]
- Ashton-Rickardt, P.G.; Murray, K. Mutants of the hepatitis B virus surface antigen that define some antigenically essential residues in the immunodominant a region. J. Med. Virol. 1989, 29, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Purdy, M.A. Hepatitis B virus S gene escape mutants. Asian J. Transfus. Sci. 2007, 1, 62–70. [Google Scholar] [CrossRef]
- Zhu, H.L.; Li, X.; Li, J.; Zhang, Z.H. Genetic variation of occult hepatitis B virus infection. World J. Gastroenterol. 2016, 22, 3531–3546. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Weinberger, K.M.; Gehrke, R.; Isogawa, M.; Hilken, G.; Kemper, T.; Xu, Y.; Yang, D.; Jilg, W.; Roggendorf, M.; et al. Mutant hepatitis B virus surface antigens (HBsAg) are immunogenic but may have a changed specificity. Virology 2004, 329, 454–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pourkarim, M.R.; Sharifi, Z.; Soleimani, A.; Amini-Bavil-Olyaee, S.; Elsadek Fakhr, A.; Sijmons, S.; Vercauteren, J.; Karimi, G.; Lemey, P.; Maes, P.; et al. Evolutionary analysis of HBV „S” antigen genetic diversity in Iranian blood donors: A nationwide study. J. Med. Virol. 2014, 86, 144–155. [Google Scholar] [CrossRef]
- Zuckerman, J.N. Protective efficacy, immunotherapeutic potential, and safety of hepatitis B vaccines. J. Med. Virol. 2006, 78, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, P.; Cerruti, R.; Icardi, G.; Bruzzone, B. Overview of hepatitis B virus mutations and their implications in the management of infection. World J. Gastroenterol. 2016, 22, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liao, Y.; Chen, J.; Cai, B.; Su, Z.; Ying, B.; Lu, X.; Tao, C.; Wang, L. Epidemiology study of HBV genotypes and antiviral drug resistance in multi-ethnic regions from Western China. Sci. Rep. 2015, 5, 17413. [Google Scholar] [CrossRef] [Green Version]
- Karimi, A.; Salimzadeh, L.; Bagheri, N. Hepatitis B virus genotyping among chronic hepatitis B individuals with resistance to Lamivudine in shahrekord, iran. Jundishapur J. Microbiol. 2014, 7, 10196. [Google Scholar] [CrossRef] [Green Version]
- Alvarado-Esquivel, C.; de la Ascensión Carrera-Gracia, M.; Conde-González, C.J.; Juárez-Figueroa, L.; Ruiz-Maya, L.; Aguilar-Benavides, S.; Torres-Valenzuela, A.; Sablon, E. Genotypic resistance to lamivudine among hepatitis B virus isolates in Mexico. J. Antimicrob. Chemother. 2006, 57, 221–223. [Google Scholar] [CrossRef]
- Mahabadi, M.; Norouzi, M.; Alavian, S.M.; Samimirad, K.; Azad, T.M.; Saberfar, E.; Mahmoodi, M.; Ramezani, F.; Karimzadeh, H.; Malekzadeh, R.; et al. Drug-related mutational patterns in hepatitis B virus (HBV) reverse transcriptase proteins from Iranian treatment-naïve chronic HBV patients. Hepat. Mon. 2013, 13, 6712. [Google Scholar] [CrossRef] [Green Version]
- Amini-Bavil-Olyaee, S.; Hosseini, S.Y.; Sabahi, F.; Alavian, S.M. Hepatitis B virus (HBV) genotype and YMDD motif mutation profile among patients infected with HBV and untreated with lamivudine. Int. J. Infect. Dis. 2008, 12, 83–87. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.-L.; Dienstag, J.; Schiff, E.; Leung, N.W.Y.; Atkins, M.; Hunt, C.; Brown, N.; Woessner, M.; Boehme, R.; Condreay, L. Prevalence and Clinical Correlates of YMDD Variants during Lamivudine Therapy for Patients with Chronic Hepatitis B. Clin. Infect. Dis. 2003, 36, 687–696. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Xu, Z.; Wang, Y.; Li, X.; Liu, L.; Chen, L.; Xin, S.; Xu, D. rtM204Q May Serve as a Novel Lamivudine-Resistance-Associated Mutation of Hepatitis B Virus. PLoS ONE 2014, 9, 89015. [Google Scholar] [CrossRef] [PubMed]
- Ismail, S.; Hafez, H.A.; Darweesh, S.K.; Kamal, K.H.; Esmat, G. Virologic response and breakthrough in chronic hepatitis B Egyptian patients receiving lamivudine therapy. Ann. Gastroenterol. 2014, 27, 380–386. [Google Scholar] [PubMed]
- Ghandehari, F.; Pourazar, A.; Zadeh, M.S.; Tajedin, N. Probing rate of YMDD motif mutant in lamivudine treatment of Iranian patients with chronic hepatitis B virus infection. Asian J. Transfus. Sci. 2011, 5, 32–34. [Google Scholar] [CrossRef] [PubMed]
- Pourkarim, M.R.; Amini-Bavil-Olyaee, S.; Lemey, P.; Maes, P.; Van Ranst, M. Are hepatitis B virus „subgenotypes” defined accurately? J. Clin. Virol. 2010, 47, 356–360. [Google Scholar] [CrossRef]
- Datta, S.; Banerjee, A.; Chandra, P.K.; Chakravarty, R. Selecting a genetic region for molecular analysis of hepatitis B virus transmission. J. Clin. Microbiol. 2007, 45, 688. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Country (Total Number of HBV Sequences) 1 | The Most Common HBV Genotype/SGT 4 (n, %) | Other SGTs |
|---|---|---|
| Algeria (4) | D4 (n = 2, 50.0%) | D2 (n = 1, 25.0%), G (n = 1, 25.0%) |
| Egypt (93) | D1 (n = 34, 36.6%) | D2 (n = 12, 12.9%), G (n = 12, 12.9%), D3 (n = 8, 8.6%), E (n = 7, 7.5%), B1 (n = 4, 4.3%), F1 (n = 3, 3.2%), F2 (n = 12, 12.9%), D7 (n = 2, 2.2%), A2 (n = 2, 2.2%), H (n = 2, 2.2%), B2 (n = 1, 1.1%), B4 (n = 1, 1.1%), C1 (n = 1, 1.1%), C2 (n = 1, 1.1%) |
| Iraq (38) | D1 (n = 30, 79.0%) | E (n = 3, 7.9%), D3 (n = 2, 5.3%), D7 (n = 2, 5.3%), G (n = 1, 2.6%) |
| Iran (2094) | D1 (n = 1958, 93.5%) | D2 (n = 52, 2.5%), D7 (n = 41, 2.0%), D3 (n = 40, 1.9%), A1 (n = 2, 0.1%), A2 (n = 52, 2.5%) |
| Jordan (37) | D1 (n = 37, 100.0%) | - |
| Kuwait (37) | D1 (n = 23, 62.2%) | D2 (n = 6, 16.2%), A1 (n = 3, 8.1%), C2 (n = 2, 5.4%), A2 (n = 1, 2.7%), E (n = 1, 2.7%), G (n = 1, 2.7%) |
| Lebanon (43) | D1 (n = 36, 83.7%) | D2 (n = 7, 16.3%) |
| Libya (2) | D1 and E (n = 1, 50.0%) | - |
| Morocco (107) | D7 (n = 46, 43.0%) | D1 (n = 17, 15.9%), A2 (n = 14, 13.1%), G (n = 12, 11.2%), H (n = 10, 9.3%), E (n = 4, 3.7%), D2 (n = 3, 2.8%), F2 (n = 1, 0.9%) |
| Northern Cyprus (69) | D1 (n = 49, 71.0%) | E (n = 8, 11.6%), A1 (n = 5, 7.2%), D2 (n = 4, 5.8%), A2 (n = 2, 2.9%), D3 (n = 1, 1.4%) |
| Oman (146) | D1 (n = 79, 54.1%) | D7 (n = 32, 21.9%), A1 (n = 30, 20.6%), D2 (n = 2, 1.4%), E (n = 1, 0.7%), C1 (n = 1, 0.7%), C2 (n = 1, 0.7%) |
| Palestine (44) | D1 (n = 39, 88.6%) | A2 (n = 3, 6.8%), D2 (n = 1, 2.3%), D3 (n = 1, 2.3%) |
| KSA 2 (501) | D1 (n = 155, 30.9%) | D7 (n = 147, 29.3%), B4 (n = 51, 10.2%), C2 (n = 36, 7.2%), C4 (n = 31, 6.2%), D2 (n = 19, 3.8%), G (n = 18, 3.6%), E (n = 16, 3.2%), C1 (n = 7, 1.4%), F2 (n = 6, 1.2%), D3 (n = 3, 0.6%), A1 (n = 3, 0.6%), B2 (n = 3, 0.6%), C3 (n = 3, 0.6%), H (n = 2, 0.4%), A2 (n = 1, 0.2%) |
| Sudan (168) | E (n = 68, 40.5%) | D1 (n = 47, 28.0%), D2 (n = 21, 12.5%), D7 (n = 19, 11.3%), D3 (n = 6, 3.6%), D4 (n = 147, 29.3%), A1 (n = 4, 2.4%), B4 (n = 1, 0.6%), H (n = 1, 0.6%), A2 (n = 1, 0.6%) |
| Somalia (98) | A1 (n = 72, 73.5%) | D7 (n = 23, 23.5%), E (n = 1, 1.0%), D1 (n = 1, 1.0%), A2 (n = 1, 1.0%) |
| Syria (75) | D1 (n = 71, 94.7%) | D2 (n = 3, 4.0%), D3 (n = 1, 1.3%) |
| Tunisia (395) | D7 (n = 176, 44.6%) | D1 (n = 149, 37.7%), F2 (n = 18, 4.6%), H (n = 17, 4.3%), G (n = 15, 3.8%), A2 (n = 8, 2.0%), D2 (n = 4, 1.0%), C2 (n = 3, 0.8%), E (n = 1, 0.3%), C4 (n = 1, 0.3%), B2 (n = 1, 0.3%) |
| Turkey (267) | D1 (n = 222, 83.2%) | G (n = 14, 5.2%), D2 (n = 12, 4.5%), D3 (n = 11, 4.1%), D7 (n = 3, 1.1%), C3 (n = 2, 0.5%), H (n = 1, 0.4%), A2 (n = 1, 0.4%), C2 (n = 1, 0.4%), E (n = 1, 0.4%), B3 (n = 1, 0.4%) |
| UAE 3 (89) | D1 (n = 58, 65.2%) | A1 (n = 16, 18.0%), D7 (n = 9, 10.1%), D2 (n = 3, 3.4%), C2 (n = 2, 2.2%), A2 (n = 1, 1.1%) |
| Yemen (33) | D1 (n = 15, 45.5%) | A1 (n = 6, 18.2%), D2 (n = 6, 18.2%), D7 (n = 6, 18.2%) |
| Antiviral Drug 1 | Trait | n 4 (%) |
|---|---|---|
| Lamivudine | S 2 | 2610 (95.5) |
| R 3 | 124 (4.5) | |
| Adefovir | S | 2919 (99.2) |
| R | 23 (0.8) | |
| Entecavir | S | 2609 (96.2) |
| R | 103 (3.8) | |
| Tenofovir | S | 2941 (99.8) |
| R | 7 (0.2) | |
| Telbivudine | S | 2635 (95.7) |
| R | 117 (4.3) |
| Antiviral Drug | Trait | Region | p Value 4 | Time of Sequence Collection | p Value | ||
|---|---|---|---|---|---|---|---|
| Middle East | North Africa | 2001–2010 | 2011–2019 | ||||
| n 3 (%) | n (%) | n (%) | n (%) | ||||
| Lamivudine | S 1 | 1956 (94.3) | 654 (99.1) | <0.001 | 1712 (95.6) | 898 (95.2) | 0.666 |
| R 2 | 118 (5.7) | 6 (0.9) | 79 (4.4) | 45 (4.8) | |||
| Adefovir | S | 2268 (99.2) | 651 (99.2) | 0.948 | 1858 (99.1) | 1061 (99.3) | 0.557 |
| R | 18 (0.8) | 5 (0.8) | 16 (0.9) | 7 (0.7) | |||
| Entecavir | S | 2136 (95.5) | 473 (99.6) | <0.001 | 1636 (96.1) | 973 (96.4) | 0.630 |
| R | 101 (4.5) | 2 (0.4) | 67 (3.9) | 36 (3.6) | |||
| Tenofovir | S | 2285 (99.8) | 656 (99.5) | 0.192 | 1869 (99.6) | 1072 (100.0) | 0.045 |
| R | 4 (0.2) | 3 (0.5) | 7 (0.4) | 0 | |||
| Telbivudine | S | 1978 (94.6) | 657 (99.4) | <0.001 | 1724 (95.9) | 911 (95.5) | 0.628 |
| R | 113 (5.4) | 4 (0.6) | 74 (4.1) | 43 (4.5) | |||
| Antiviral Drug | Mutation n 1 (%) |
|---|---|
| Lamivudine | 204I (n = 39); 180M,204I (n = 15); 180M,204V (n = 15); 173L,180M,204V (n = 8); 204I,80I (n = 6); 180M (n = 5); 180M,204I,80I (n = 5); 173Q,180G,204S,181T (n = 4); 180M,204V,80V (n = 4); 180M,204I,80V (n = 3); 173L,180M,204I,181T (n = 2); 181T (n = 2); 204I,80V (n = 2); 173A,180F,204E,181T,80H (n = 1); 173I,180R,204A,181T (n = 1); 173L,180M (n = 1); 173L,180M,204I,80I (n = 1); 173L,180P,204G,181T (n = 1); 173P,180P,204I (n = 1); 173R,180R,204P,181V,80H (n = 1); 173R,180R,204S,181V,80H (n = 1); 173W,180D,204L,181T (n = 1); 180M,204V,181V (n = 1); 181T,181S (n = 1); 181V (n = 1); 204S (n = 1); 204V (n = 1) |
| Adefovir | 181T (n = 8); 236T (n = 3); 181T,236A (n = 2); 181V (n = 2); 181E,236T (n = 1); 181I,236T (n = 1); 181P,236T (n = 1); 181T,181S (n = 1); 181T,236L (n = 1); 181T,236T (n = 1); 181V,236Q (n = 1); 181V,236V (n = 1) |
| Entecavir | 204I (n = 47); 204V,180M (n = 25); 204I,180M (n = 24); 184A,204V,180M (n = 1); 184S,204I,180M (n = 1); 202I,169G,184W,250C,204I,180P (n = 1); 202I,204I,180M (n = 1); 202I,204V,180M (n = 1); 204V (n = 1); 204V,202C,180M (n = 1) |
| Tenofovir | 236T (n = 7) |
| Telbivudine | 204I (n = 55); 204V (n = 24); 204I,80I (n = 12); 204I,80V (n = 5); 204R,181T (n = 4); 204V,80V (n = 4); 181T (n = 2); 204I,181T (n = 2); 181T,181S (n = 1); 181V (n = 1); 204A,181T (n = 1); 204E,80H,181T (n = 1); 204G,181T (n = 1); 204L,181T (n = 1); 204P,80H,181V (n = 1); 204S,80H,181V (n = 1); 204V,181V (n = 1) |
| Genotype/SGT 1 | Number of the MENA Sequences Analyzed 2 | Number of Sequences within Clusters (%) | Dyads 3 | Small Networks 4 | Large Networks 5 |
|---|---|---|---|---|---|
| D1 | 1777 | 267 (15.0) | Iran (31), Syria (3), Tunisia (1), Turkey (1), Iran and Oman (1) | Size 3: Iran (4), KSA (1), Tunisia (1), Egypt and KSA (1), Iran, Sudan and Syria (1); Size 4: Iran (7), Iran and Turkey (2), Iran and Sudan (1), Iran, Lebanon and Oman (1); Size 5: Iran (2); Size 6: Iran (2); Size 7: Iran (2); Size 8: Iran, Morocco, Lebanon, Turkey and Tunisia; Size 11: Iran (1); Size 12: Iran (1); Size 13: Iran and Turkey (1); Size 14: Iran (2) | Size 31: Iran, Oman, Palestine, Sudan, Tunisia, Turkey (1) |
| D7 | 268 | 166 (61.9) | Tunisia (4), Morocco and Tunisia (3), Somalia (2), Sudan and Tunisia (1) | Size 3: Morocco, Oman and Tunisia (1), Oman, Somalia and Tunisia (1), Tunisia (1); Size 4: Tunisia (2), Morocco and Tunisia (1); Size 6: Somalia and Tunisia; Size 7: Algeria, Morocco, Oman, Sudan and Tunisia (1), Morocco, Oman and Tunisia (1); Size 8: Morocco, Oman and Tunisia (1), Morocco, Sudan and Tunisia (1); Size 9: Morocco, Somalia and Tunisia (1), Oman (1), Oman, Sudan and Tunisia (1); Size 13: Algeria, Morocco, Sudan and Tunisia (1) | Size 15: Morocco and Tunisia (1), Morocco and Tunisia (1); Size 17: Morocco, Sudan and Tunisia (1); Size 31: Iran and Oman (1) |
| A1 | 102 | 0 | |||
| D2 | 78 | 22 (28.2) | Iran (4), Sudan (1), Syria (1) | Size 5: Iran (1), Iran and Lebanon (1) | |
| E | 73 | 18 (24.7) | Sudan (1) | Size 4: Egypt and Sudan (1); Size 14: Sudan (1) | |
| D3 | 48 | 4 (8.3) | Size 4: Iran (1) | ||
| A2 | 23 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Athamneh, R.Y.; Arıkan, A.; Sayan, M.; Mahafzah, A.; Sallam, M. Variable Proportions of Phylogenetic Clustering and Low Levels of Antiviral Drug Resistance among the Major HBV Sub-Genotypes in the Middle East and North Africa. Pathogens 2021, 10, 1333. https://doi.org/10.3390/pathogens10101333
Athamneh RY, Arıkan A, Sayan M, Mahafzah A, Sallam M. Variable Proportions of Phylogenetic Clustering and Low Levels of Antiviral Drug Resistance among the Major HBV Sub-Genotypes in the Middle East and North Africa. Pathogens. 2021; 10(10):1333. https://doi.org/10.3390/pathogens10101333
Chicago/Turabian StyleAthamneh, Rabaa Y., Ayşe Arıkan, Murat Sayan, Azmi Mahafzah, and Malik Sallam. 2021. "Variable Proportions of Phylogenetic Clustering and Low Levels of Antiviral Drug Resistance among the Major HBV Sub-Genotypes in the Middle East and North Africa" Pathogens 10, no. 10: 1333. https://doi.org/10.3390/pathogens10101333