
Vascular Changes and Hypoxia in Periodontal Disease as a Link to Systemic Complications


{kind=link}
{kind=link}
Abstract
:1. Introduction: Overview of the Vascular Impact of Periodontal and Oral Inflammation
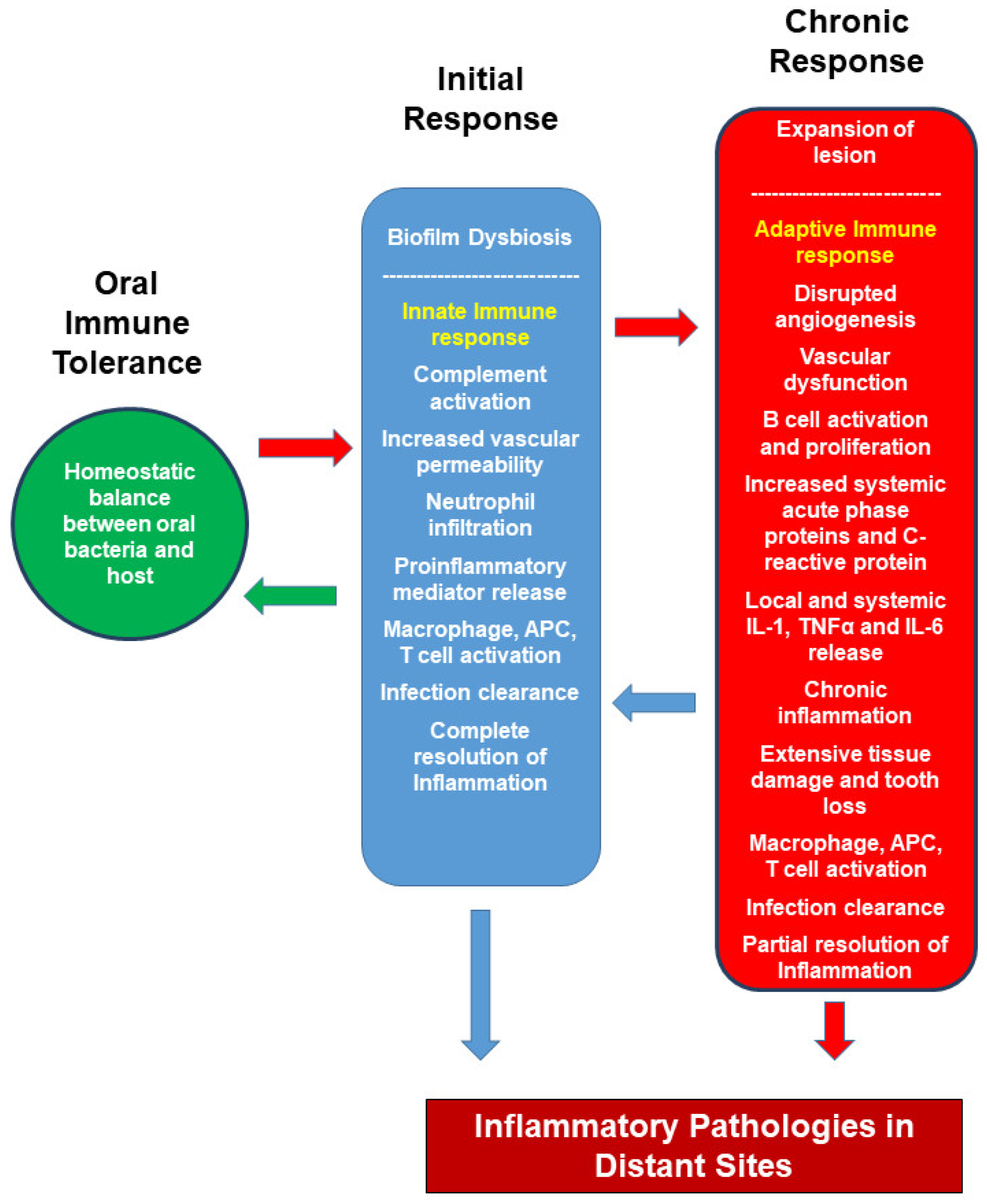
2. Host Immune Response to Oral Bacteria
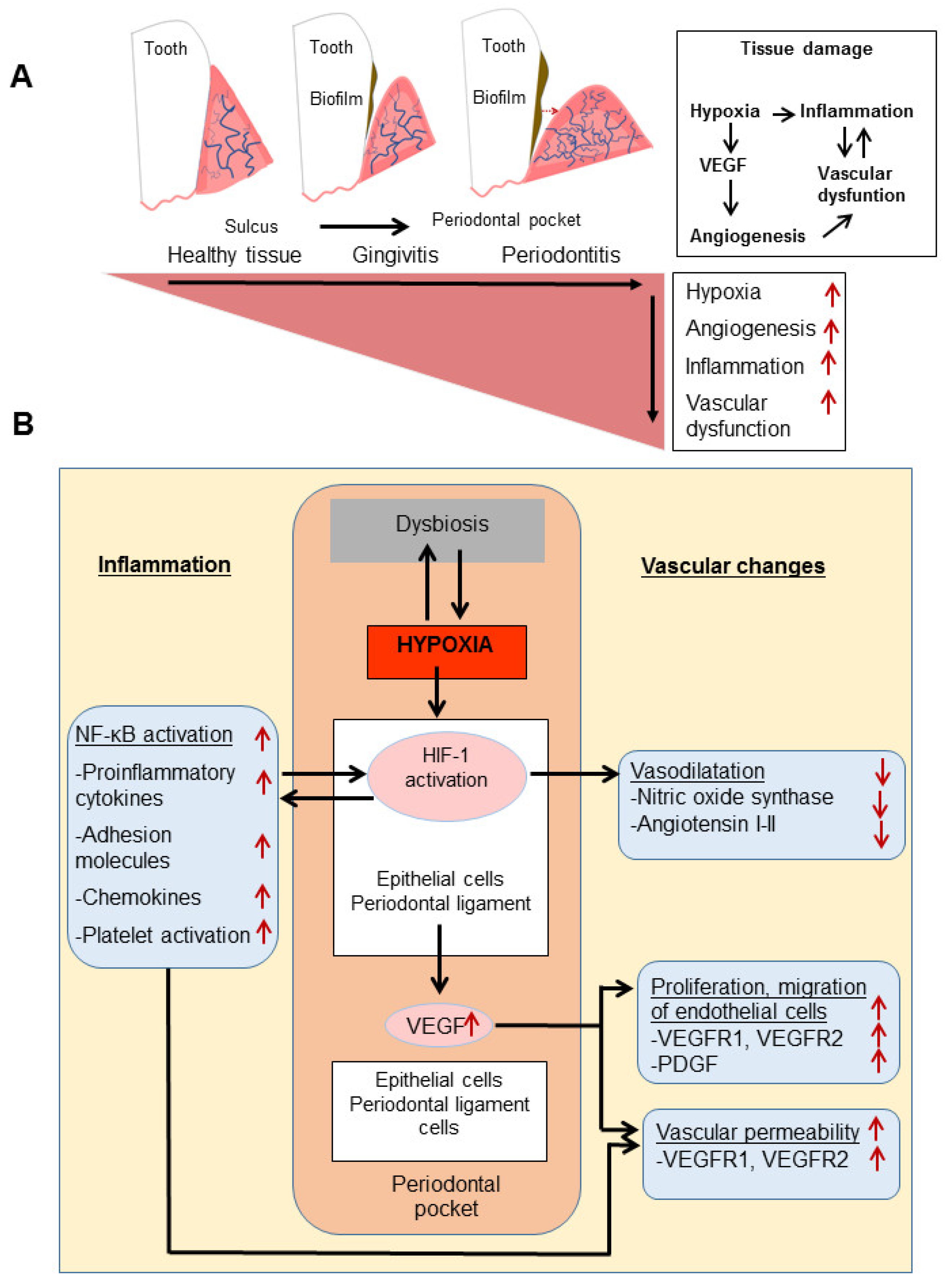
3. Local Inflammation and Vascular Changes
4. Hypoxia in Local Inflammation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hajishengallis, G. Periodontitis: From microbial immune subversion to systemic inflammation. Nat. Rev. 2015, 15, 30–44. [Google Scholar] [CrossRef]
- Methé, B.A.; Nelson, K.E.; Pop, M.; Creasy, H.H.; Giglio, M.G.; Huttenhower, C.; Gevers, D.; Petrosino, J.; Abubucker, S.; Badger, J.; et al. A framework for human microbiome research. Nature 2012, 486, 215–221. [Google Scholar] [CrossRef]
- Belkaid, Y.; Artis, D. Immunity at the Barriers. Eur. J. Immunol. 2013, 43, 3096–3100. [Google Scholar] [CrossRef] [Green Version]
- Eke, P.I.; Page, R.C.; Wei, L.; Thornton-Evans, G.; Genco, R.J. Update of the case definitions for population-based surveillance of periodontitis. J. Periodontol. 2012, 83, 1449–1454. [Google Scholar] [CrossRef]
- Bui, F.Q.; Almeida-da-Silva, C.L.C.; Huynh, B.; Trinh, A.; Liu, J.; Woodward, J.; Asadi, H.; Ojcius, D.M. Association between periodontal pathogens and systemic diseas. Biomed. J. 2019, 42, 27–35. [Google Scholar] [CrossRef]
- Preshaw, P.M.; Alba, A.L.; Herrera, D.; Jepsen, S. Periodontitis and diabetes: A two-way relationship. Diabetologia 2012, 55, 21–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naguib, G.; Al-mashat, H.; Desta, T.; Graves, D.T. Diabetes Prolongs the Inflammatory Response to a Bacterial Stimulus Through Cytokine Dysregulation. J. Investig. Dermatol. 2004, 123, 87–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, S.; Takahashi, S.-S.; Tokutomi, F.-A.; Yoshida, A.; Kobayashi, K.; Yoshino, F.; Wada-Takahashi, S.; Toyama, T.; Watanabe, K.; Hamada, N.; et al. Gingival vascular functions are altered in type 2 diabetes mellitus model and/or periodontitis model. J. Clin. Biochem. Nutr. 2012, 51, 108–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrugia, C.; Stafford, G.P.; Potempa, J.; Wilkinson, R.N.; Chen, Y. Mechanisms of vascular damage by systemic dissemination of the oral pathogen Porphyromonas gingivalis. FEBS J. 2021, 288, 1479–1495. [Google Scholar] [CrossRef] [PubMed]
- Amar, S.; Gokce, N.; Morgan, S.; Loukideli, M.; Van Dyke, T.E.; Vita, J.A. Periodontal Disease Is Associated With Brachial Artery Endothelial Dysfunction and Systemic Inflammation. Arter. Thromb Vasc Biol. 2003, 23, 1245–1249. [Google Scholar] [CrossRef] [Green Version]
- Koizumi, Y.; Oguchi, S.; Yamamoto, M. Nasal Immunization with Porphyromonas gingivalis Outer Membrane Protein Decreases P. gingivalis -Induced Atherosclerosis and Inflammation in Spontaneously Hyperlipidemic Mice. Infect. Immun. 2008, 76, 2958–2965. [Google Scholar] [CrossRef] [Green Version]
- Fiehn, N.; Larsen, T.; Christiansen, N.; Holmstrup, P.; Schroeder, T.V. Identification of Periodontal Pathogens in Atherosclerotic Vessels. J. Periodontol. 2005, 76, 731–736. [Google Scholar] [CrossRef]
- Mahendra, J.; Mahendra, L.; Kurian, V.M.; Jaishankar, K.; Mythilli, R. Prevalence of periodontal pathogens in coronary atherosclerotic plaque of patients undergoing coronary artery bypass graft surgery. J. Maxillofac Oral Surg. 2009, 8, 108–113. [Google Scholar] [CrossRef] [Green Version]
- Miyakawa, H.; Honma, K.; Qi, M.; Kuramitsu, H.K. Interaction of Porphyromonas gingivalis with low-density lipoproteins: Implications for a role for periodontitis in atherosclerosis. J. Periodontal Res. 2003, 39, 1–9. [Google Scholar] [CrossRef]
- Ren, X.Y.; Wang, M.M.; Zhao, Y.; Wang, C.; Shi, X.X.; Gao, J.H. Effects of Periodontal Intervention on Levels of Serum High- sensitivity C-reactive Protein and Interleukin 6, and on Carotid Artery in Rats with Chronic Periodontitis and Hyperlipidemia. Chin. J. Dent. Res. 2019, 22, 203–209. [Google Scholar] [CrossRef]
- Galea, J.; Armstrong, J.; Gadsdon, P.; Holden, H.; Francis, S.E.; Holt, M. Interleukin-1β in Coronary Arteries of Patients With Ischemic Heart Disease. Arter. Thromb. Vasc. Bio. 1996, 16, 1000–1006. [Google Scholar] [CrossRef]
- Barath, P.; Fishbein, M.C.; Cao, J.; Berenson, J.; Helfant, R.H.; Forrester, J.S. Detection and Localization of Tumor Necrosis Factor in Human Atheroma. Am. J. Cardiol. 1990, 65, 297–302. [Google Scholar] [CrossRef]
- Loos, B.G.; Craandijk, J.; Hoek, F.J.; Wertheim-van Dillen, P.M.E.; Van Der Velden, U. Elevation of Systemic Markers Related to Cardiovascular Diseases in the Peripheral Blood of Periodontitis Patients. J. Periodontol. 2000, 71, 1528–1534. [Google Scholar] [CrossRef] [PubMed]
- Duarte, P.M.; da Rocha, M.; Sampaio, E.; Mestnik, M.J.; Feres, M.; Figueiredo, L.C.; Bastos, M.F.; Faveri, M. Serum Levels of Cytokines in Subjects With Generalized Chronic and Aggressive Periodontitis Before and After Non-Surgical Periodontal Therapy: A Pilot Study. J. Periodontol. 2010, 81, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- Slade, G.D.; Ghezzi, E.M.; Heiss, G.; Beck, J.D.; Riche, E.; Offenbacher, S. Relationship between periodontal disease and C-reactive protein among adults in the atherosclerosis risk in communities study. Arch. Intern. Med. 2003, 163, 1172–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suh, J.S.; Lee, S.H.; Fouladian, Z.; Lee, J.Y.; Kim, T.; Kang, M.K.; Lusis, A.J.; Boström, K.I.; Kim, R.H.; Park, N.H. Rosuvastatin Prevents the Exacerbation of Atherosclerosis in Ligature-Induced Periodontal Disease Mouse Model. Sci. Rep. 2020, 10, 6383–6399. [Google Scholar] [CrossRef] [Green Version]
- Van Dyke, T.E.; Hasturk, H.; Abdallah, R.; Kantarci, A.; Nguyen, D.; Giordano, N.; Hamilton, J. Resolvin, E1 (RvE1) attenuates atherosclerotic plaque Formation in diet and inflammation-induced atherogenesis. Arter. Thromb Vasc Biol. 2015, 35, 1123–1133. [Google Scholar] [CrossRef] [Green Version]
- Ceccarelli, F.; Saccucci, M.; Carlo, G.D.; Lucchetti, R.; Pilloni, A.; Pranno, N.; Luzzi, V.; Valesini, G.; Polimeni, A. Periodontitis and rheumatoid arthritis: The same inflammatory mediators? Mediat. Inflamm. 2019, 2019, 6034546. [Google Scholar] [CrossRef]
- Zhou, M.; Li, S.; Pathak, J.L. Pro-inflammatory cytokines and osteocytes. Curr. Osteoporos. Rep. 2019, 17, 97–104. [Google Scholar] [CrossRef]
- Totaro, M.C.; Cattani, P.; Ria, F.; Tolusso, B.; Gremese, E.; Fedele, A.L.; D’Onghia, S.; Marchetti, S.; Sante, G.D.; Canestri, S.; et al. Porphyromonas gingivalis and the pathogenesis of rheumatoid arthritis: Analysis of various compartments including the synovial tissue. Arthritis Res. Ther. 2013, 15, R66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichert, S.; Haffner, M.; Keyßer, G.; Schäfer, C.; Stein, J.M.; Schaller, H.G.; Wienke, A.; Strauss, H.; Heide, S.; Schulz, S. Detection of oral bacterial DNA in synovial fluid. J. Clin. Periodontol. 2013, 40, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Gully, N.; Bright, R.; Marino, V.; Marchant, C.; Cantley, M.; Haynes, D.; Butler, C.; Dashper, S.; Reynolds, E.; Bartold, M. Porphyromonas gingivalis peptidylarginine deiminase, a key contributor in the pathogenesis of experimental periodontal disease and experimental arthritis. PLoS ONE 2014, 9, e100838. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, Y.; Ouhara, K.; Munenaga, S.; Shoji, M.; Ozawa, T.; Hisatsune, J.; Kado, I.; Kajiya, M.; Matsuda, S.; Kawai, T.; et al. Effect of Porphyromonas gingivalis infection on gut dysbiosis and resultant arthritis exacerbation in mouse model. Arthritis Res Ther. 2020, 22, 249. [Google Scholar] [CrossRef] [PubMed]
- Courbon, G.; Rinaudo-Gaujous, M.; Blasco-Baque, V.; Auger, I.; Caire, R.; Mijola, L.; Vico, L.; Paul, S.; Marotte, H. Porphyromonas gingivalis experimentally induces periodontis and an anti-CCP2-associated arthritis in the rat. Ann. Rheum. Dis. 2019, 78, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Konkel, J.E.; O’Boyle, C.; Krishnan, S. Distal consequences of oral inflammation. Front. Immunol. 2019, 10, 1403. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, K.; Wegner, N.; Yucel-Lindberg, N.; Venables, P.J. Periodontitis in RA-the citrullinated enolase connection. Nat. Rev. Rheumatol. 2010, 6, 727–730. [Google Scholar] [CrossRef]
- Liao, F.; Li, Z.; Wang, Y.; Shi, B.; Gong, Z.; Cheng, X. Porphyromonas gingivalis may play an important role in the pathogenesis of periodontitis-associated rheumatoid arthritis. Med. Hypotheses 2009, 72, 732–735. [Google Scholar] [CrossRef] [PubMed]
- Lamster, I.; Sonis, S.; Hannigan, A.; Kolodkin, A. An association between Crohn’s disease, periodontal disease and enhanced neutrophil function. J. Periodontol 1978, 49, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Van Dyke, T.E.; Dowell, V.R.; Offenbacher, S.; Snyder, W.; Hersh, T. Potential role of microorganisms isolated from periodontal lesions in the pathogenesis of inflammatory bowel disease. Infect. Immun. 1986, 53, 671–677. [Google Scholar] [CrossRef] [Green Version]
- She, Y.-Y.; Kong, X.-B.; Ge, Y.-P.; Liu, Z.-Y.; Chen, J.-Y.; Jiang, J.-W.; Jiang, H.B.; Fang, S.L. Periodontitis and inflammatory bowel disease: A meta-analysis. BMC Oral Health 2020, 20, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strauss, J.; Kaplan, G.G.; Beck, P.L.; Rioux, K.; Panaccione, R.; Devinney, R.; Lynch, T.; Allen-Vercoe, E. Invasive potential of gut mucosa-derived Fusobacterium nucleatum positively correlates with IBD status of the host. Inflamm Bowel Dis. 2011, 17, 1971–1978. [Google Scholar] [CrossRef] [PubMed]
- Kitamoto, S.; Nagao-Kitamoto, H.; Hein, R.; Schmidt, T.M.; Kamada, N. The Bacterial Connection between the Oral Cavity and the Gut Diseases. J. Dent. Res. 2020, 99, 1021–1029. [Google Scholar] [CrossRef]
- Kitamoto, S.; Nagao-Kitamoto, H.; Jiao, Y.; Gillilland, M.G.; Hayashi, A.; Imai, J.; Sugihara, K.; Miyoshi, M.; Brazil, J.C.; Kuffa, P.; et al. The Intermucosal Connection between the Mouth and Gut in Commensal Pathobiont-Driven Colitis. Cell 2020, 182, 447–462. [Google Scholar] [CrossRef]
- Said, H.S.; Suda, W.; Nakagome, S.; Chinen, H.; Oshima, K.; Kim, S.; Kimura, R.; Iraha, A.; Ishida, H.; Fujita, J.; et al. Dysbiosis of salivary microbiota in inflammatory bowel disease and its association with oral immunological biomarkers. DNA Res. 2014, 21, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.; Yamazaki, K.; Nakajima, M.; Date, Y.; Kikuchi, J.; Hase, K.; Ohno, H.; Yamazaki, K. Oral Administration of Porphyromonas gingivalis Alters the Gut Microbiome and Serum Metabolome. mSphere 2018, 3, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huh, J.W.; Roh, T.Y. Opportunistic detection of Fusobacterium nucleatum as a marker for the early gut microbial dysbiosis. BMC Microbiol. 2020, 20, 1–17. [Google Scholar] [CrossRef]
- Rubinstein, M.R.; Baik, J.E.; Lagana, S.M.; Han, R.P.; Raab, W.J.; Sahoo, D.; Dalerba, P.; Wang, T.C.; Han, Y.W. Fusobacterium nucleatum promotes colorectal cancer by inducing Wnt/β-catenin modulator Annexin A1. EMBO Rep. 2019, 20, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kanagasingam, S.; Chukkapalli, S.S.; Welbury, R.; Singhrao, S.K. Porphyromonas gingivalis is a Strong Risk Factor for Alzheimer’s Disease. J. Alzheimer’s Dis. Rep. 2020, 4, 501–511. [Google Scholar] [CrossRef]
- Chen, C.K.; Wu, Y.T.; Chang, Y.C. Association between chronic periodontitis and the risk of Alzheimer’s disease: A retrospective, population-based, matched-cohort study. Alzheimer’s Res. Ther. 2017, 9, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Damgaard, C.; Kantarci, A.; Holmstrup, P.; Hasturk, H.; Nielsen, C.H.; Van Dyke, T.E. Porphyromonas gingivalis-induced production of reactive oxygen species, TNF-α, IL-6, CXCL8 and CCL2 by neutrophils from localized aggressive periodontitis and healthy donors Modulating actions of red blood cells and resolvin E1. J. Periodontal Res. 2017, 52, 246–254. [Google Scholar] [CrossRef] [Green Version]
- Singer, R.E.; Moss, K.; Kim, S.J.; Beck, J.D.; Offenbacher, S. Oxidative stress and IgG antibody modify periodontitis-CRP association. J. Dent. Res. 2015, 94, 1698–1705. [Google Scholar] [CrossRef] [Green Version]
- Kantarci, A.; Tognoni, C.M.; Yaghmoor, W.; Marghalani, A.; Stephens, D.; Ahn, J.Y.; Carreras, I.; Dedeoglu, A. Microglial response to experimental periodontitis in a murine model of Alzheimer’s disease. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ilievski, V.; Zuchowska, P.K.; Green, S.J.; Toth, P.T.; Ragozzino, M.E.; Le, K.; Aljewari, H.W.; O’Brien-Simpson, N.M.; Reynolds, E.C.; Watanabe, K. Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and amyloid beta production in wild type mice. PLoS ONE 2018, 13, e0204941. [Google Scholar] [CrossRef] [Green Version]
- Cekici, A.; Kantarci, A.; Hasturk, H.; Van Dyke, T.E. Inflammatory and immune pathways in the pathogenesis of periodontal disease. Periodontol. 2000 2014, 64, 57–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Pablo, P.; Chapple, I.L.C.; Buckley, C.; Dietrich, T. Periodontitis in systemic rheumatic diseases. Nat. Rev. Rheumatol. 2009, 5, 218–224. [Google Scholar] [CrossRef]
- Sanz, M.; Marco del Castillo, A.; Jepsen, S.; Gonzalez-Juanatey, J.R.; D’Aiuto, F.; Bouchard, P.; Chapple, I.; Dietrich, T.; Gotsman, I.; Graziani, F.; et al. Periodontitis and cardiovascular diseases: Consensus report. J. Clin. Periodontol. 2020, 47, 268–288. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, S.; Hagner, M.; Nogueira, A.V.B.; Franke, A.; Jäger, A.; Deschner, J. Inflammatory bowel disease and oral health:systematic review and a meta-analysis. J. Clin. Periodontol. 2017, 44, 382–393. [Google Scholar] [CrossRef] [Green Version]
- Sulijaya, B.; Takahashi, N.; Yamazaki, K. Host modulation therapy using anti-inflammatory and antioxidant agents in periodontitis: A review to a clinical translation. Arch. Oral Biol. 2019, 105, 72–80. [Google Scholar] [CrossRef]
- Van Dyke, T.E.; Kantarci, A.; Hasturk, H. Host-mediated resolution of inflammation in periodontal diseases. Periodontol. 2000 2006, 40, 144–163. [Google Scholar]
- Humphrey, S.; Williamson, R. Saliva Review. J. Prosthet Dent. 2001, 85, 162–169. [Google Scholar] [CrossRef]
- Delima, A.; Van Dyke, T.E. Origin and function of the cellular components in gingival crevice fluid. Periodontol. 2000 2003, 31, 55–76. [Google Scholar] [CrossRef] [PubMed]
- Dutzan, N.; Konkel, J.; Greenwell-Wild, T.; Moutsopoulos, N. Characterization of the human immune cell network at the gingival barrier. Mucosal Immunol. 2016, 9, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Chena, X.; Chena, L.; Tan, J.; Shi, D.; Ke, T.; Leia, L. Th17 and Th1 Lymphocytes Are Correlated with Chronic Periodontitis. Immunol. Investig. 2016, 45, 243–254. [Google Scholar] [CrossRef]
- Suárez, L.; Vargas, D.; Rodríguez, A.; Arce, R.; Roa, N. Systemic Th17 response in the presence of periodontal inflammation Abstract. J. Appl Oral Sci. 2020, 28, 1–7. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Tapping, R.I.; Harokopakis, E.; Nishiyama, S.; Pukar, R.; Schifferle, R.E.; Lyle, E.A.; Triantafilou, M.; Triantafilou, K.; Yoshimura, F. Differential interactions of fimbriae and lipopolysaccharide from Porphyromonas gingivalis with the Toll-like receptor 2-centred pattern recognition apparatus. Cell. Microbiol. 2006, 8, 1557–1570. [Google Scholar] [CrossRef]
- Lambris, J.; Hajishengallis, G. Microbial manipulation of receptor cross-talk in innate immunity. Nat. Rev. Immunol. 2011, 11, 187–200. [Google Scholar] [CrossRef] [Green Version]
- Hajishengallis, G.; Maekawa, T.; Abe, T.; Hajishengallis, E.; Lambris, J. Complement involvement in periodontitis: Molecular mechanisms and rational therapeutic approaches. Adv. Exp. Med. Biol. 2015, 865, 57–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selders, G.; Fetz, A.; Radic, M.; Bowlin, G.L. An overview of the role of neutrophils in innate immunity, inflammation and host-biomaterial integration. Regen. Biomater. 2017, 4, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.A.; Krauss, J.L. Neutrophils in periodontal inflammation. Periodontal Dis. 2011, 15, 56–83. [Google Scholar] [CrossRef] [Green Version]
- Upadhyay, J.; Upadhyay, R.B.; Agrawal, P.; Jaitley, S.; Shekhar, R. Langerhans cells and their role in oral mucosal diseases. N. Am. J. Med Sci. 2013, 5, 505–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manakil, J. Periodontal Diseases—A Clinician’s Guide; Books on Demand: Norderstedt, Germany, 2012. [Google Scholar]
- Yu, T.; Zhao, L.; Huang, X.; Ma, C.; Wang, Y.; Zhang, J.; Xuan, D. Enhanced Activity of the Macrophage M1/M2 Phenotypes and Phenotypic Switch to M1 in Periodontal Infection. J. Periodontol. 2016, 87, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Ohlrich, E.J.; Cullinan, M.P.; Seymour, G.J. The immunopathogenesis of periodontal disease. Aust. Dent. J. 2009, 54, S2–S10. [Google Scholar] [CrossRef]
- Noguchi, K.; Miwa, Y.; Sunohara, M.; Sato, I. Analysis of vascular distribution and growth factors in human gingival tissue associated with periodontal probing depth. Okajimas Folia Anat. Jpn. 2011, 88, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Melincovici, C.; Al, E. Vascular endothelial growth factor (VEGF)—key factor in normal and pathological angiogenesis. Rom. J. Morphol. Embryo. 2018, 59, 455–467. [Google Scholar]
- Tischer, E.; Michelle, R.; Hartman, T.; Silva, M.; Gospodarowicz, D.; Fiddes, J.C.; Abraham, J.A. The human gene for vascular endothelial growth factor: Multiple protein forms are encoded through alternative exon splicing. J. Biol. Chem. 1991, 266, 11947–11954. [Google Scholar] [CrossRef]
- Shaik-Dasthagirisaheb, Y.B.; Varvara, G.; Murmura, G.; Saggini, A.; Potalivo, G.; Caraffa, A.; Antinolfi, P.; Tetè, S.; Tripodi, D.; Conti, F.; et al. Vascular endothelial growth factor (VEGF), mast cells and inflammation. Int. J. Immunopathol. Pharmacol. 2013, 26, 327–335. [Google Scholar] [CrossRef]
- McLaughlin, A.; De Vries, G. Role of PLCγ and Ca2+ in VEGF- and FGF-induced choroidal endothelial cell proliferation. Am. J. Physiol.-Cell Physiol. 2001, 281, 1448–1456. [Google Scholar] [CrossRef]
- Angelo, L.S.; Kurzrock, R. Vascular endothelial growth factor and its relationship to inflammatory mediators. Clin. Cancer Res. 2007, 13, 2825–2830. [Google Scholar] [CrossRef] [Green Version]
- Akagi, Y.; Liu, W.; Xie, K.; Zebrowski, B.; Shaheen, R.; Ellis, L. Regulation of vascular endothelial growth factor expression in human colon cancer by interleukin-1β. Br. J. Cancer 1999, 80, 1506–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, H.H.; Lai, W.W.; Chen, H.H.W.; Liu, H.S.; Su, W.C. Autocrine IL-6-induced Stat3 activation contributes to the pathogenesis of lung adenocarcinoma and malignant pleural effusion. Oncogene 2006, 25, 4300–4309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.H.; Kirschenbaum, A.; Lu, M.; Yao, S.; Dosoretz, A.; Holland, J.F.; Levine, A.C. Prostaglandin E2 induces hypoxia-inducible factor-1α stabilization and nuclear localization in a human prostate cancer cell line. J. Biol. Chem. 2002, 277, 50081–50086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McColl, B.K.; Stacker, S.A.; Achen, M.G. Molecular regulation of the VEGF family - Inducers of angiogenesis and lymphangiogenesis. Apmis 2004, 112, 463–480. [Google Scholar] [CrossRef]
- Zoellner, H.; Chapple, C.C.; Hunter, N. Microvasculature in gingivitis and chronic periodontitis: Disruption of vascular networks with protracted inflammation. Microsc. Res. Tech. 2002, 56, 15–31. [Google Scholar] [CrossRef]
- Nassar, H.; Chou, H.; Khlgatian, M.; Iii, F.C.G.; Van Dyke, T.E.; Genco, C.A. Role for Fimbriae and Lysine-Specific Cysteine Proteinase Gingipain K in Expression of Interleukin-8 and Monocyte Chemoattractant Protein in Porphyromonas gingivalis -Infected Endothelial Cells. Infect. Immun. 2002, 70, 268–276. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Davey, M.; Yumoto, H.; Iii, F.C.G.; Genco, C.A. Fimbria-dependent activation of pro-inflammatory molecules in Porphyromonas gingivalis infected human aortic endothelial cells. Cell. Microbiol. 2006, 8, 738–757. [Google Scholar] [CrossRef]
- Walter, C.; Zahlten, J.; Schmeck, B.; Schaudinn, C.; Hippenstiel, S.; Frisch, E.; Hocke, A.C.; Pischon, N.; Kuramitsu, H.K.; Bernimoulin, J.-P.; et al. Porphyromonas gingivalis Strain-Dependent Activation of Human Endothelial Cells. Infect. Immun. 2004, 72, 5910–5918. [Google Scholar] [CrossRef] [Green Version]
- Yun, P.L.W.; Decarlo, A.A.; Hunter, N. Gingipains of Porphyromonas gingivalis Modulate Leukocyte Adhesion Molecule Expression Induced in Human Endothelial Cells by Ligation of CD99. Infect. Immun. 2006, 74, 1661–1672. [Google Scholar] [CrossRef] [Green Version]
- Imamura, T. The Role of Gingipains in the Pathogenesis of Periodontal Disease. J. Periodontol. 2003, 74, 111–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sima, A.V.; Stancu, C.S.; Simionescu, M. Vascular endothelium in atherosclerosis. Cell Tissue Res. 2009, 335, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Barneo, J.; Pardal, R.; Ortega-Saenz, P. Cellular Mechanisms of Oxygen Sensing. Annu. Rev. Physiol. 2001, 63, 259–287. [Google Scholar] [CrossRef] [PubMed]
- Humar, R.O.K.; Kiefer, F.N.; Berns, H. Hypoxia enhances vascular cell proliferation and angiogenesis in vitro via rapamycin ( mTOR ) -dependent signaling. FASEB J. 2002, 16, 771–780. [Google Scholar] [CrossRef] [Green Version]
- Devraj, G.; Beerlage, C.; Brüne, B.; Kempf, V.A.J. Hypoxia and HIF-1 activation in bacterial infections. Microbes Infect. 2017, 19, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Chiu, P.; Hsu, P.; Lin, S.; Peng, I.; Wang, C. Pathophysiological implications of hypoxia in human diseases. J. Biomed. Sci. 2020, 27, 63. [Google Scholar] [CrossRef]
- Ahluwalia, A.; Tarnawski, A.S. Critical Role of Hypoxia Sensor—HIF-1 in VEGF Gene Activation. Implications for Angiogenesis and Tissue Injury Healing. Curr Med. Chem. 2012, 19, 90–97. [Google Scholar] [CrossRef]
- Heikal, L.; Ghezzi, P.; Mengozzi, M.; Stelmaszczuk, B.; Feelisch, M.; Ferns, G. Erythropoietin and a nonerythropoietic peptide analog promote aortic endothelial cell repair under hypoxic conditions: Role of nitric oxide. Hypoxia 2016, 4, 121–133. [Google Scholar] [CrossRef] [Green Version]
- Heikal, L.; Ghezzi, P.; Mengozzi, M.; Ferns, G. Low Oxygen Tension Primes Aortic Endothelial Cells to the Reparative Effect of Tissue-Protective Cytokines. Mol. Med. 2015, 21, 709–716. [Google Scholar] [CrossRef]
- Van Uden, P.; Kenneth, N.S.; Webster, R.; Müller, H.A.; Mudie, S.; Rocha, S. Evolutionary conserved regulation of HIF-1β by NFκB. PLoS Genet. 2011, 7, e0204941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frede, S.; Stockmann, C.; Freitag, P.; Fandrey, J. Bacterial lipopolysaccharide induces HIF-1 activation in human monocytes via p44/42 MAPK and NF-κB. Biochem. J. 2006, 396, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-P.; Li, F.Y.L.; Xu, A.; Cheng, B.; Tsao, S.W.; Fung, M.-L.; Leung, W.K. Lipopolysaccharide and Hypoxia-Induced HIF-1 Activation in Human Gingival Fibroblasts. J. Periodontol 2012, 83, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Gölz, L.; Memmert, S.; Rath-Deschner, B.; Jäger, A.; Appel, T.; Baumgarten, G.; Götz, W.; Frede, S. Hypoxia and P. gingivalis synergistically induce HIF-1 and NF-κB activation in PDL cells and periodontal diseases. Mediat. Inflamm. 2015, 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, W.; Shi, J.; Zhao, Y.; Yan, F.; Lei, L.; Li, H. Porphyromonas gingivalis triggers inflammatory responses in periodontal ligament cells by succinate-succinate dehydrogenase–HIF–1α axis. Biochem. Biophys. Res. Commun. 2020, 522, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Bui, F.Q.; Johnson, L.; Roberts, J.; Hung, S.C.; Lee, J.; Atanasova, K.R.; Huang, P.R.; Yilmaz, O.; Ojcius, D.M. Fusobacterium nucleatum infection of gingival epithelial cells leads to NLRP3 inflammasome-dependent secretion of IL-1β and the danger signals ASC and HMGB1. Cell Microbiol. 2016, 18, 970–981. [Google Scholar] [CrossRef] [Green Version]
- Görlach, A.; Diebold, I.; Schini-Kerth, V.B.; Berchner-Pfannschmidt, U.; Roth, U.; Brandes, R.P.; Kietzmann, T.; Busse, R. Thrombin activates the hypoxia-inducible factor-1 signaling pathway in vascular smooth muscle cells role of the p22phox-containing NADPH oxidase. Circ. Res. 2001, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, H.; Makino, T.; Okamoto, K.; Poellinger, L.; Ohnuma, K.; Morimoto, C.; Tanaka, H. TCR Engagement Increases Hypoxia-Inducible Factor-1α Protein Synthesis via Rapamycin-Sensitive Pathway under Hypoxic Conditions in Human Peripheral T Cells. J. Immunol. 2005, 174, 7592–7599. [Google Scholar] [CrossRef] [Green Version]
- Santos, C.; Akashi, A.; Dionísio, T.; Sipert, C.; Didier, D.; Greene, A.; Oliveira, S.; Pereira, H.; Becari, C.; Oliveira, E.; et al. Characterization of a Local Renin-Angiotensin System in Rat Gingival Tissue. J. Periodontol 2009, 80, 130–139. [Google Scholar] [CrossRef] [Green Version]
- Santos, C.F.; Morandini, A.C.; Dionisio, T.; de Faria, F.A.C.; Lima, M.C.; Figueiredo, C.M.; Colombini-Ishikiriama, B.L.; Sipert, C.; Maciel, R.P.; Akashi, A.P.; et al. Functional local renin-angiotensin system in human and rat periodontal tissue. PLoS ONE 2015, 10, e0134601. [Google Scholar] [CrossRef]
- Janjić, K.; Schellner, A.; Engenhart, A.; Kernstock, K.; Schädl, B.; Moritz, A.; Agis, H. Angiopoietin-like 4 production upon treatment with hypoxia and L-mimosine in periodontal fibroblasts. J. Periodontal Res. 2019, 54, 489–498. [Google Scholar] [CrossRef]
- Proctor, D.M.; Shelef, K.M.; Gonzalez, A.; Davis, C.L.; Dethlefsen, L.; Burns, A.R.; Loomer, P.M.; Armitage, G.C.; Ryder, M.I.; Millman, M.E.; et al. Microbial biogeography and ecology of the mouth and implications for periodontal diseases. Periodontol. 2020, 2000, 26–41. [Google Scholar] [CrossRef]
- Mendes, R.T.; Nguyen, D.; Stephens, D.; Pamuk, F.; Van Dyke, T.E.; Fernandes, D.; Kantarci, A. Endothelial cell response to Fusobacterium nucleatum. Infect. Immun. 2016, 84, 2141–2148. [Google Scholar] [CrossRef] [Green Version]
- Aruni, A.W.; Dou, Y.; Mishra, A.; Fletcher, H.M. The Biofilm Community: Rebels with a Cause. Curr. Oral Health Rep. 2015, 2, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Xu, S.; Zhao, H.; Liu, L.; Lv, X.; Hu, F. Hypoxia and Porphyromonas gingivalis-lipopolysaccharide synergistically induce NLRP3 inflammasome activation in human gingival fibroblasts. Int. Immunopharmacol. 2020, 94. [Google Scholar] [CrossRef]
- Cheng, R.; Liu, W.; Zhang, R.; Feng, Y.; Bhowmick, N.A.; Hu, T. Porphyromonas gingivalis-Derived Lipopolysaccharide Combines Hypoxia to Induce Caspase-1 Activation in Periodontitis. Front. Cell Infect. Microbiol. 2017, 14, 474. [Google Scholar] [CrossRef]
- Mendes, R.T.; Nguyen, D.; Stephens, D.; Pamuk, F.; Hasturk, H.; Van Dyke, T.E.; Fernandes, D.; Kantarci, A. Hypoxia-induced endothelial cell responses-possible roles during periodontal disease. Clin. Exp. Dent. Res. 2018, 14, 241–248. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Celik, D.; Kantarci, A. Vascular Changes and Hypoxia in Periodontal Disease as a Link to Systemic Complications. Pathogens 2021, 10, 1280. https://doi.org/10.3390/pathogens10101280
Celik D, Kantarci A. Vascular Changes and Hypoxia in Periodontal Disease as a Link to Systemic Complications. Pathogens. 2021; 10(10):1280. https://doi.org/10.3390/pathogens10101280
Chicago/Turabian StyleCelik, Dilek, and Alpdogan Kantarci. 2021. "Vascular Changes and Hypoxia in Periodontal Disease as a Link to Systemic Complications" Pathogens 10, no. 10: 1280. https://doi.org/10.3390/pathogens10101280