An Integrated Approach to Identify New Anti-Filarial Leads to Treat River Blindness, a Neglected Tropical Disease

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Experimental Screening to Identify Active Macrofilaricidal Compounds Against Adult B. pahangi
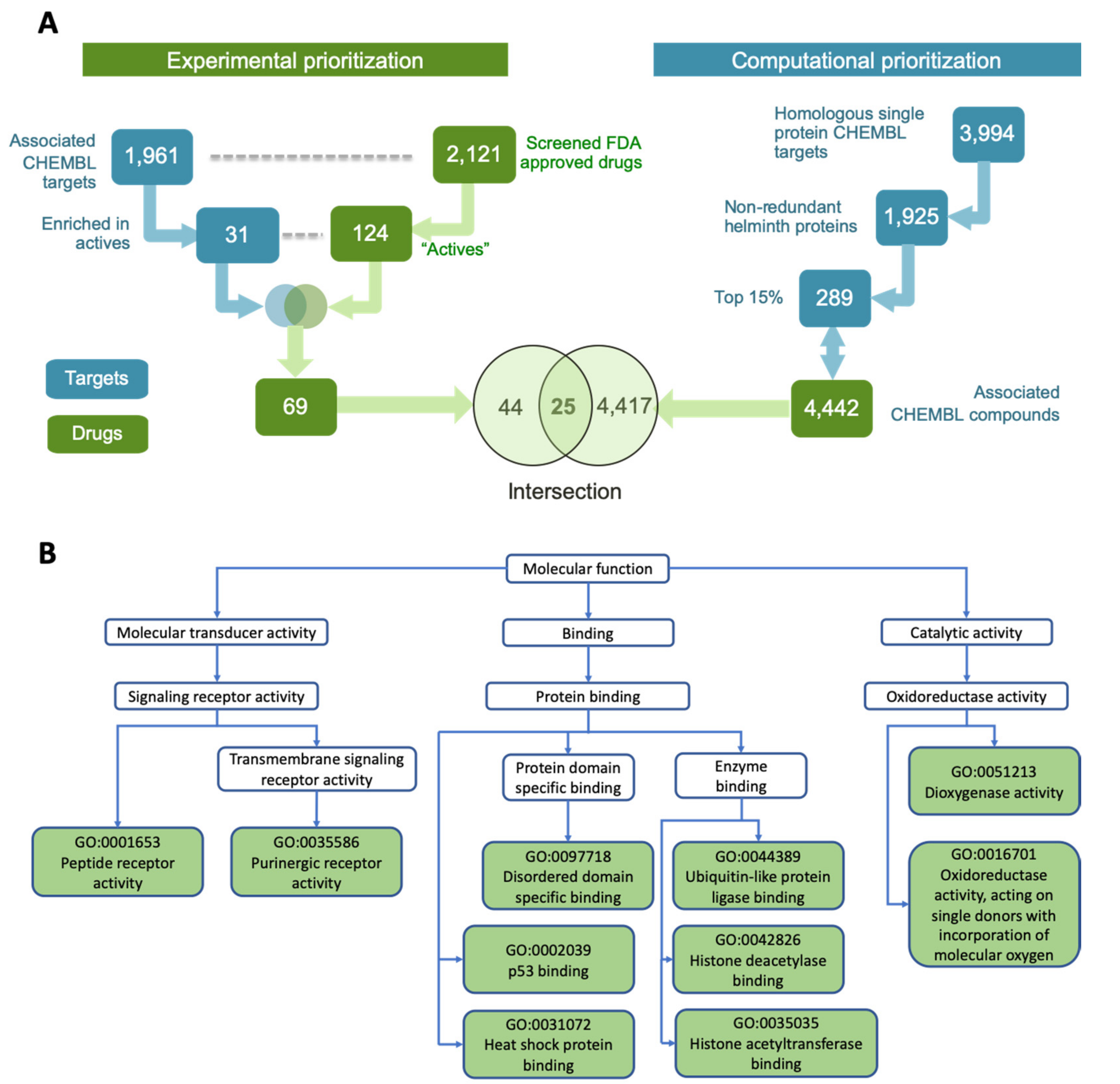
2.2. Integrating Active Drugs with Computational Prioritization
2.3. Screening of Prioritized Hits in Other Filarial Species
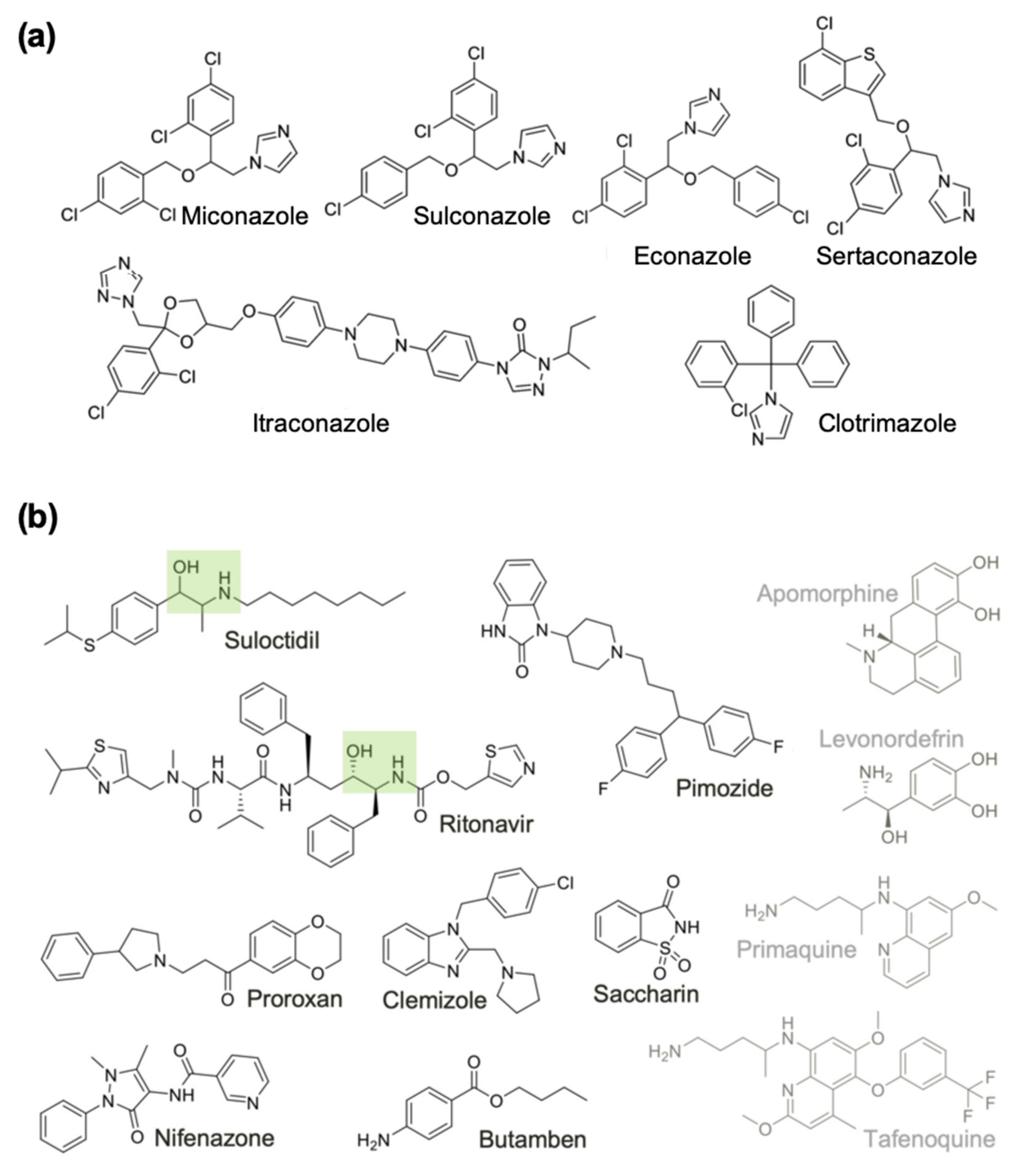
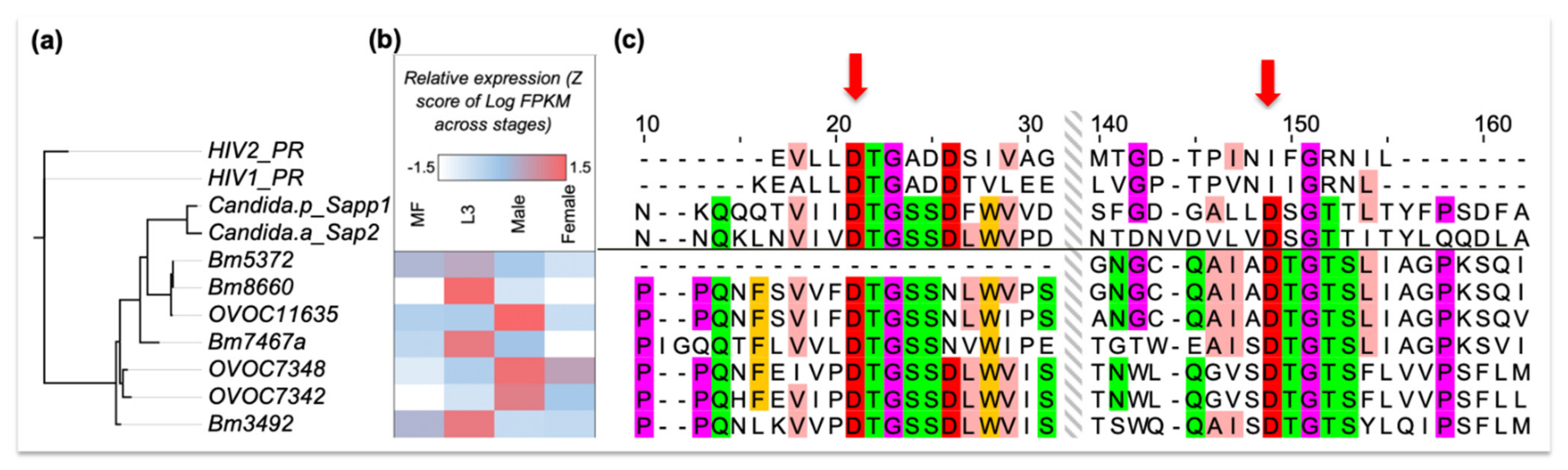
2.4. Expanding the List of Potential Anti-Macrofilarial Drugs by Focusing on Two Specific Classes
3. Conclusions
4. Materials and Methods
4.1. Ethics Statement
4.2. Experimental Screening of a Library of Drugs Approved for Clinical Use Against B. pahangi
4.3. Experimental Screening of Prioritized Drugs in O. ochengi Adults
4.4. Counter Screening Compounds with L. loa Microfilariae in Vitro
4.5. O. volvulus L5 Motility and Viability Assay
4.6. O. volvulus L4 Motility Assay
4.7. Computational Identification and Prioritization of Targets and Inhibitors
4.8. Expanding the List of Azoles as Specific Drug Class with Anti-Macrofilarial Potency
4.9. Expanding the List of Aspartic Protease Inhibitors
4.10. Clustering of Hits Based on Structural Similarity
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Taylor, M.J.; Hoerauf, A.; Bockarie, M. Lymphatic filariasis and onchocerciasis. Lancet 2010, 376, 1175–1185. [Google Scholar] [CrossRef]
- Hoerauf, A.; Pfarr, K.; Mand, S.; Debrah, A.Y.; Specht, S. Filariasis in Africa-treatment challenges and prospects. Clin. Microbiol. Infect. 2011, 17, 977–985. [Google Scholar] [CrossRef] [Green Version]
- Lustigman, S.; Prichard, R.K.; Gazzinelli, A.; Grant, W.N.; Boatin, B.A.; McCarthy, J.S.; Basanez, M.G. A research agenda for helminth diseases of humans: The problem of helminthiases. PLoS Negl. Trop. Dis. 2012, 6, e1582. [Google Scholar] [CrossRef] [Green Version]
- Prichard, R.K.; Basanez, M.G.; Boatin, B.A.; McCarthy, J.S.; Garcia, H.H.; Yang, G.J.; Sripa, B.; Lustigman, S. A research agenda for helminth diseases of humans: Intervention for control and elimination. PLoS Negl. Trop. Dis. 2012, 6, e1549. [Google Scholar] [CrossRef] [Green Version]
- King, C.L.; Suamani, J.; Sanuku, N.; Cheng, Y.C.; Satofan, S.; Mancuso, B.; Goss, C.W.; Robinson, L.J.; Siba, P.M.; Weil, G.J.; et al. A Trial of a Triple-Drug Treatment for Lymphatic Filariasis. N. Engl. J. Med. 2018, 379, 1801–1810. [Google Scholar] [CrossRef]
- Weil, G.J.; Bogus, J.; Christian, M.; Dubray, C.; Djuardi, Y.; Fischer, P.U.; Goss, C.W.; Hardy, M.; Jambulingam, P.; King, C.L.; et al. The safety of double- and triple-drug community mass drug administration for lymphatic filariasis: A multicenter, open-label, cluster-randomized study. PLoS Med. 2019, 16, e1002839. [Google Scholar] [CrossRef] [Green Version]
- Irvine, M.A.; Stolk, W.A.; Smith, M.E.; Subramanian, S.; Singh, B.K.; Weil, G.J.; Michael, E.; Hollingsworth, T.D. Effectiveness of a triple-drug regimen for global elimination of lymphatic filariasis: A modelling study. Lancet Infect. Dis. 2017, 17, 451–458. [Google Scholar] [CrossRef] [Green Version]
- Milton, P.; Hamley, J.I.D.; Walker, M.; Basanez, M.G. Moxidectin: An oral treatment for human onchocerciasis. Expert Rev. Anti Infect. Ther. 2020, 18, 1067–1081. [Google Scholar] [CrossRef] [PubMed]
- Molyneux, D.H.; Bradley, M.; Hoerauf, A.; Kyelem, D.; Taylor, M.J. Mass drug treatment for lymphatic filariasis and onchocerciasis. Trends Parasitol. 2003, 19, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Molyneux, D.H.; Taylor, M.J. Current status and future prospects of the Global Lymphatic Filariasis Programme. Curr. Opin. Infect. Dis. 2001, 14, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Molyneux, D.H.; Hopkins, A.; Bradley, M.H.; Kelly-Hope, L.A. Multidimensional complexities of filariasis control in an era of large-scale mass drug administration programmes: A can of worms. Parasites Vectors 2014, 7, 363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaisier, A.P.; van Oortmarssen, G.J.; Remme, J.; Habbema, J.D. The reproductive lifespan of Onchocerca volvulus in West African savanna. Acta Trop. 1991, 48, 271–284. [Google Scholar] [CrossRef] [Green Version]
- WHO. Global programme to eliminate lymphatic filariasis. Wkly Epidemiol. Rec. 2010, 85, 365–372. [Google Scholar]
- Ottesen, E.A.; Hooper, P.J.; Bradley, M.; Biswas, G. The global programme to eliminate lymphatic filariasis: Health impact after 8 years. PLoS Negl. Trop. Dis. 2008, 2, e317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, B.K.; Hooper, P.J.; Bradley, M.H.; McFarland, D.A.; Ottesen, E.A. The economic benefits resulting from the first 8 years of the Global Programme to Eliminate Lymphatic Filariasis (2000–2007). PLoS Negl. Trop. Dis. 2010, 4, e708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diawara, L.; Traore, M.O.; Badji, A.; Bissan, Y.; Doumbia, K.; Goita, S.F.; Konate, L.; Mounkoro, K.; Sarr, M.D.; Seck, A.F.; et al. Feasibility of onchocerciasis elimination with ivermectin treatment in endemic foci in Africa: First evidence from studies in Mali and Senegal. PLoS Negl. Trop. Dis. 2009, 3, e497. [Google Scholar] [CrossRef] [Green Version]
- Traore, M.O.; Sarr, M.D.; Badji, A.; Bissan, Y.; Diawara, L.; Doumbia, K.; Goita, S.F.; Konate, L.; Mounkoro, K.; Seck, A.F.; et al. Proof-of-principle of onchocerciasis elimination with ivermectin treatment in endemic foci in Africa: Final results of a study in Mali and Senegal. PLoS Negl. Trop. Dis. 2012, 6, e1825. [Google Scholar] [CrossRef] [Green Version]
- NTD Modelling Consortium Onchocerciasis Group. The World Health Organization 2030 goals for onchocerciasis: Insights and perspectives from mathematical modelling: NTD Modelling Consortium Onchocerciasis Group. Gates Open Res. 2019, 3, 1545. [Google Scholar] [CrossRef]
- NTD Modelling Consortium Lymphatic Filariasis Group. The roadmap towards elimination of lymphatic filariasis by 2030: Insights from quantitative and mathematical modelling. Gates Open Res. 2019, 3, 1538. [Google Scholar] [CrossRef] [Green Version]
- Turner, H.C.; Churcher, T.S.; Walker, M.; Osei-Atweneboana, M.Y.; Prichard, R.K.; Basanez, M.G. Uncertainty surrounding projections of the long-term impact of ivermectin treatment on human onchocerciasis. PLoS Negl. Trop. Dis. 2013, 7, e2169. [Google Scholar] [CrossRef] [Green Version]
- Turner, H.C.; Walker, M.; Churcher, T.S.; Osei-Atweneboana, M.Y.; Biritwum, N.K.; Hopkins, A.; Prichard, R.K.; Basanez, M.G. Reaching the London Declaration on Neglected Tropical Diseases Goals for Onchocerciasis: An Economic Evaluation of Increasing the Frequency of Ivermectin Treatment in Africa. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2014, 59, 923–932. [Google Scholar] [CrossRef] [PubMed]
- GBD 2015 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1545–1602. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, A.D. Neglected tropical diseases in Africa: A new paradigm. Int. Health 2016, 8 (Suppl. 1), i28–i33. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.E.; Remme, J.H.; Steinmann, P.; Stolk, W.A.; Roungou, J.B.; Tediosi, F. Control, elimination, and eradication of river blindness: Scenarios, timelines, and ivermectin treatment needs in Africa. PLoS Negl. Trop. Dis. 2015, 9, e0003664. [Google Scholar] [CrossRef] [Green Version]
- African Programme for Onchocerciasis Control (APOC). Report of the Thirty-Eight Session of the Technical Consultative Committee: Ouagadougou; Document Number: DIR/COORD/APOC/REP/TCC38; WHO: Geneva, Switzerland, 2014. [Google Scholar]
- The U.S. Food and Drug Administration (FDA); The United States Department of Agriculture (USDA). Drug Approval Package: Moxidectin; The U.S. Food and Drug Administration (FDA): Silver Springs, MD, USA, 2018.
- Awadzi, K.; Opoku, N.O.; Attah, S.K.; Lazdins-Helds, J.; Kuesel, A.C. A randomized, single-ascending-dose, ivermectin-controlled, double-blind study of moxidectin in Onchocerca volvulus infection. PLoS Negl. Trop. Dis. 2014, 8, e2953. [Google Scholar] [CrossRef] [PubMed]
- Opoku, N.O.; Bakajika, D.K.; Kanza, E.M.; Howard, H.; Mambandu, G.L.; Nyathirombo, A.; Nigo, M.M.; Kasonia, K.; Masembe, S.L.; Mumbere, M.; et al. Single dose moxidectin versus ivermectin for Onchocerca volvulus infection in Ghana, Liberia, and the Democratic Republic of the Congo: A randomised, controlled, double-blind phase 3 trial. Lancet 2018, 392, 1207–1216. [Google Scholar] [CrossRef] [Green Version]
- Turner, H.C.; Walker, M.; Attah, S.K.; Opoku, N.O.; Awadzi, K.; Kuesel, A.C.; Basanez, M.G. The potential impact of moxidectin on onchocerciasis elimination in Africa: An economic evaluation based on the Phase II clinical trial data. Parasites Vectors 2015, 8, 167. [Google Scholar] [CrossRef] [Green Version]
- Kelly-Hope, L.A.; Cano, J.; Stanton, M.C.; Bockarie, M.J.; Molyneux, D.H. Innovative tools for assessing risks for severe adverse events in areas of overlapping Loa loa and other filarial distributions: The application of micro-stratification mapping. Parasites Vectors 2014, 7, 307. [Google Scholar] [CrossRef] [Green Version]
- Boussinesq, M.; Gardon, J.; Gardon-Wendel, N.; Chippaux, J.P. Clinical picture, epidemiology and outcome of Loa-associated serious adverse events related to mass ivermectin treatment of onchocerciasis in Cameroon. Filaria J. 2003, 2, S4. [Google Scholar] [CrossRef] [Green Version]
- D’Ambrosio, M.V.; Bakalar, M.; Bennuru, S.; Reber, C.; Skandarajah, A.; Nilsson, L.; Switz, N.; Kamgno, J.; Pion, S.; Boussinesq, M.; et al. Point-of-care quantification of blood-borne filarial parasites with a mobile phone microscope. Sci. Transl. Med. 2015, 7, 286re284. [Google Scholar] [CrossRef] [Green Version]
- Boatin, B.A.; Basanez, M.G.; Prichard, R.K.; Awadzi, K.; Barakat, R.M.; Garcia, H.H.; Gazzinelli, A.; Grant, W.N.; McCarthy, J.S.; N’Goran, E.K.; et al. A research agenda for helminth diseases of humans: Towards control and elimination. PLoS Negl. Trop. Dis. 2012, 6, e1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osei-Atweneboana, M.Y.; Awadzi, K.; Attah, S.K.; Boakye, D.A.; Gyapong, J.O.; Prichard, R.K. Phenotypic evidence of emerging ivermectin resistance in Onchocerca volvulus. PLoS Negl. Trop. Dis. 2011, 5, e998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, S.R.; Bourguinat, C.; Nana-Djeunga, H.C.; Kengne-Ouafo, J.A.; Pion, S.D.S.; Bopda, J.; Kamgno, J.; Wanji, S.; Che, H.; Kuesel, A.C.; et al. Genome-wide analysis of ivermectin response by Onchocerca volvulus reveals that genetic drift and soft selective sweeps contribute to loss of drug sensitivity. PLoS Negl. Trop. Dis. 2017, 11, e0005816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lustigman, S.; McCarter, J.P. Ivermectin Resistance in Onchocerca volvulus: Toward a Genetic Basis. PLoS Negl. Trop. Dis. 2007, 1, e76. [Google Scholar] [CrossRef] [Green Version]
- Debrah, A.Y.; Specht, S.; Klarmann-Schulz, U.; Batsa, L.; Mand, S.; Marfo-Debrekyei, Y.; Fimmers, R.; Dubben, B.; Kwarteng, A.; Osei-Atweneboana, M.; et al. Doxycycline Leads to Sterility and Enhanced Killing of Female Onchocerca volvulus Worms in an Area with Persistent Microfilaridermia After Repeated Ivermectin Treatment: A Randomized, Placebo-Controlled, Double-Blind Trial. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2015, 61, 517–526. [Google Scholar] [CrossRef] [Green Version]
- Veale, C.G.L. Unpacking the Pathogen Box-An Open Source Tool for Fighting Neglected Tropical Disease. ChemMedChem 2019, 14, 386–453. [Google Scholar] [CrossRef]
- Voronin, D.; Tricoche, N.; Jawahar, S.; Shlossman, M.; Bulman, C.A.; Fischer, C.; Suderman, M.T.; Sakanari, J.A.; Lustigman, S. Development of a preliminary in vitro drug screening assay based on a newly established culturing system for pre-adult fifth-stage Onchocerca volvulus worms. PLoS Negl. Trop. Dis. 2019, 13, e0007108. [Google Scholar] [CrossRef] [Green Version]
- International Helminth Genomes Consortium. Comparative genomics of the major parasitic worms. Nat. Genet. 2019, 51, 163–174. [Google Scholar] [CrossRef] [Green Version]
- Marcellino, C.; Gut, J.; Lim, K.C.; Singh, R.; McKerrow, J.; Sakanari, J. WormAssay: A novel computer application for whole-plate motion-based screening of macroscopic parasites. PLoS Negl. Trop. Dis. 2012, 6, e1494. [Google Scholar] [CrossRef]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Felix, E.; Magarinos, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Sheth, S.; Brito, R.; Mukherjea, D.; Rybak, L.P.; Ramkumar, V. Adenosine receptors: Expression, function and regulation. Int. J. Mol. Sci. 2014, 15, 2024–2052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldhoer, M.; Bartlett, S.E.; Whistler, J.L. Opioid receptors. Annu. Rev. Biochem. 2004, 73, 953–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef] [PubMed]
- Kerscher, O.; Felberbaum, R.; Hochstrasser, M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu. Rev. Cell Dev. Biol. 2006, 22, 159–180. [Google Scholar] [CrossRef] [Green Version]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [Green Version]
- Balamurugan, K. HIF-1 at the crossroads of hypoxia, inflammation, and cancer. Int. J. Cancer 2016, 138, 1058–1066. [Google Scholar] [CrossRef]
- Mashima, R.; Okuyama, T. The role of lipoxygenases in pathophysiology; new insights and future perspectives. Redox Biol. 2015, 6, 297–310. [Google Scholar] [CrossRef] [Green Version]
- Sheehan, D.J.; Hitchcock, C.A.; Sibley, C.M. Current and emerging azole antifungal agents. Clin. Microbiol. Rev. 1999, 12, 40–79. [Google Scholar] [CrossRef] [Green Version]
- Epstein, B.J.; Vogel, K.; Palmer, B.F. Dihydropyridine calcium channel antagonists in the management of hypertension. Drugs 2007, 67, 1309–1327. [Google Scholar] [CrossRef]
- Marella, A.; Tanwar, O.P.; Saha, R.; Ali, M.R.; Srivastava, S.; Akhter, M.; Shaquiquzzaman, M.; Alam, M.M. Quinoline: A versatile heterocyclic. Saudi Pharm. J. 2013, 21, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Deliorman, D.; Calis, I.; Ergun, F.; Dogan, B.S.; Buharalioglu, C.K.; Kanzik, I. Studies on the vascular effects of the fractions and phenolic compounds isolated from Viscum album ssp. album. J. Ethnopharmacol. 2000, 72, 323–329. [Google Scholar] [CrossRef]
- Malaisse, W.J. Calcium-antagonists and islet function X. Effect of suloctidie. Arch. Int. Pharmacodyn. Ther. 1977, 228, 339–344. [Google Scholar] [PubMed]
- Chatelain, P.; Reckinger, N.; Roncucci, R. Effect of suloctidil on Na+/K+ ATPase activity and on membrane fluidity in rat brain synaptosomes. Biochem. Pharmacol. 1979, 28, 3677–3680. [Google Scholar] [CrossRef]
- Chen, X.; Ji, Z.L.; Chen, Y.Z. TTD: Therapeutic Target Database. Nucleic Acids Res. 2002, 30, 412–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise, L.D.; Pattison, I.C.; Butler, D.E.; DeWald, H.A.; Lewis, E.P.; Lobbestael, S.J.; Nordin, I.C.; Poschel, B.P.; Coughenour, L.L. 1-[3-(Diarylamino)propyl]piperidines and related compounds, potential antipsychotic agents with low cataleptogenic profiles. J. Med. Chem. 1985, 28, 606–612. [Google Scholar] [CrossRef]
- Katagi, J.; Nakamura, Y.; Cao, X.; Ohara, H.; Honda, A.; Izumi-Nakaseko, H.; Ando, K.; Sugiyama, A. Why Can dl-Sotalol Prolong the QT Interval In Vivo Despite Its Weak Inhibitory Effect on hERG K(+) Channels In Vitro? Electrophysiological and Pharmacokinetic Analysis with the Halothane-Anesthetized Guinea Pig Model. Cardiovasc. Toxicol. 2016, 16, 138–146. [Google Scholar] [CrossRef]
- Bang, S.; Yang, T.J.; Yoo, S.; Heo, T.H.; Hwang, S.W. Inhibition of sensory neuronal TRPs contributes to anti-nociception by butamben. Neurosci. Lett. 2012, 506, 297–302. [Google Scholar] [CrossRef]
- Finkelstein, M.; Kromer, C.M.; Sweeney, S.A.; Delahunt, C.S. Some aspects of the pharmacology of clemizole hydrochloride. J. Am. Pharm. Assoc. 1960, 49, 18–22. [Google Scholar] [CrossRef]
- Richter, J.M.; Schaefer, M.; Hill, K. Clemizole hydrochloride is a novel and potent inhibitor of transient receptor potential channel TRPC5. Mol. Pharmacol. 2014, 86, 514–521. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, I.V.; Bondarenko, R.A. Individual Approach to the Use of Medications to Normalize Situational Anxiety under the Psycho-Emotional Stress. Aviakosm. Ekolog. Med. 2015, 49, 32–35. [Google Scholar]
- Mahon, B.P.; Hendon, A.M.; Driscoll, J.M.; Rankin, G.M.; Poulsen, S.A.; Supuran, C.T.; McKenna, R. Saccharin: A lead compound for structure-based drug design of carbonic anhydrase IX inhibitors. Bioorg. Med. Chem. 2015, 23, 849–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kempf, D.J.; Sham, H.L.; Marsh, K.C.; Flentge, C.A.; Betebenner, D.; Green, B.E.; McDonald, E.; Vasavanonda, S.; Saldivar, A.; Wideburg, N.E.; et al. Discovery of ritonavir, a potent inhibitor of HIV protease with high oral bioavailability and clinical efficacy. J. Med. Chem. 1998, 41, 602–617. [Google Scholar] [CrossRef] [PubMed]
- Ikezoe, T.; Hisatake, Y.; Takeuchi, T.; Ohtsuki, Y.; Yang, Y.; Said, J.W.; Taguchi, H.; Koeffler, H.P. HIV-1 protease inhibitor, ritonavir: A potent inhibitor of CYP3A4, enhanced the anticancer effects of docetaxel in androgen-independent prostate cancer cells in vitro and in vivo. Cancer Res. 2004, 64, 7426–7431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunthard, H.F.; Aberg, J.A.; Eron, J.J.; Hoy, J.F.; Telenti, A.; Benson, C.A.; Burger, D.M.; Cahn, P.; Gallant, J.E.; Glesby, M.J.; et al. Antiretroviral treatment of adult HIV infection: 2014 recommendations of the International Antiviral Society-USA Panel. JAMA 2014, 312, 410–425. [Google Scholar] [CrossRef]
- Schlessinger, A.; Geier, E.; Fan, H.; Irwin, J.J.; Shoichet, B.K.; Giacomini, K.M.; Sali, A. Structure-based discovery of prescription drugs that interact with the norepinephrine transporter, NET. Proc. Natl Acad. Sci. USA 2011, 108, 15810–15815. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Thiessen, P.A.; Cheng, T.; Yu, B.; Shoemaker, B.A.; Wang, J.; Bolton, E.E.; Wang, Y.; Bryant, S.H. Literature information in PubChem: Associations between PubChem records and scientific articles. J. Cheminform. 2016, 8, 32. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, L.; Luque-Ortega, J.R.; Manzano, J.I.; Castanys, S.; Rivas, L.; Gamarro, F. Tafenoquine, an antiplasmodial 8-aminoquinoline, targets leishmania respiratory complex III and induces apoptosis. Antimicrob. Agents Chemother. 2010, 54, 5344–5351. [Google Scholar] [CrossRef] [Green Version]
- Graves, P.R.; Kwiek, J.J.; Fadden, P.; Ray, R.; Hardeman, K.; Coley, A.M.; Foley, M.; Haystead, T.A. Discovery of novel targets of quinoline drugs in the human purine binding proteome. Mol. Pharmacol. 2002, 62, 1364–1372. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.S.; Bancone, G.; Nosten, F.; White, N.J.; Luzzatto, L. Primaquine-induced haemolysis in females heterozygous for G6PD deficiency. Malar. J. 2018, 17, 101. [Google Scholar] [CrossRef]
- Anden, N.E.; Rubenson, A.; Fuxe, K.; Hokfelt, T. Evidence for dopamine receptor stimulation by apomorphine. J. Pharm. Pharmacol. 1967, 19, 627–629. [Google Scholar] [CrossRef]
- Anden, N.E.; Strombom, U. Adrenergic receptor blocking agents: Effects on central noradrenaline and dopamine receptors and on motor activity. Psychopharmacologia 1974, 38, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Warrilow, A.G.; Parker, J.E.; Kelly, D.E.; Kelly, S.L. Azole affinity of sterol 14alpha-demethylase (CYP51) enzymes from Candida albicans and Homo sapiens. Antimicrob. Agents Chemother. 2013, 57, 1352–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, S.; Rice, C.A.; Zhang, T.; Edrada-Ebel, R.; Henriquez, F.L.; Roberts, C.W. Characterisation of sterol biosynthesis and validation of 14alpha-demethylase as a drug target in Acanthamoeba. Sci. Rep. 2017, 7, 8247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrillo-Munoz, A.J.; Tur-Tur, C.; Giusiano, G.; Marcos-Arias, C.; Eraso, E.; Jauregizar, N.; Quindos, G. Sertaconazole: An antifungal agent for the topical treatment of superficial candidiasis. Expert Rev. Anti Infect. Ther. 2013, 11, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Warrilow, A.G.; Hull, C.M.; Rolley, N.J.; Parker, J.E.; Nes, W.D.; Smith, S.N.; Kelly, D.E.; Kelly, S.L. Clotrimazole as a potent agent for treating the oomycete fish pathogen Saprolegnia parasitica through inhibition of sterol 14alpha-demethylase (CYP51). Appl. Environ. Microbiol. 2014, 80, 6154–6166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pace, J.R.; DeBerardinis, A.M.; Sail, V.; Tacheva-Grigorova, S.K.; Chan, K.A.; Tran, R.; Raccuia, D.S.; Wechsler-Reya, R.J.; Hadden, M.K. Repurposing the Clinically Efficacious Antifungal Agent Itraconazole as an Anticancer Chemotherapeutic. J. Med. Chem. 2016, 59, 3635–3649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odds, F.C.; Brown, A.J.; Gow, N.A. Antifungal agents: Mechanisms of action. Trends Microbiol. 2003, 11, 272–279. [Google Scholar] [CrossRef]
- Makepeace, B.L.; Tanya, V.N. 25 Years of the Onchocerca ochengi Model. Trends Parasitol. 2016, 32, 966–978. [Google Scholar] [CrossRef] [Green Version]
- Gardon, J.; Gardon-Wendel, N.; Demanga, N.; Kamgno, J.; Chippaux, J.P.; Boussinesq, M. Serious reactions after mass treatment of onchocerciasis with ivermectin in an area endemic for Loa loa infection. Lancet 1997, 350, 18–22. [Google Scholar] [CrossRef]
- Drugs for Neglected Diseases Initiative. Filaria: River blindness, Oxfendazole. Available online: https://dndi.org/research-development/portfolio/oxfendazole/ (accessed on 11 November 2020).
- Drugs for Neglected Diseases Initiative. Filaria: River Blindness, Emodepside. Available online: https://dndi.org/research-development/portfolio/emodepside/ (accessed on 11 November 2020).
- Hubner, M.P.; Martin, C.; Specht, S.; Koschel, M.; Dubben, B.; Frohberger, S.J.; Ehrens, A.; Fendler, M.; Struever, D.; Mitre, E.; et al. Oxfendazole mediates macrofilaricidal efficacy against the filarial nematode Litomosoides sigmodontis in vivo and inhibits Onchocerca spec. motility in vitro. PLoS Negl. Trop. Dis. 2020, 14, e0008427. [Google Scholar] [CrossRef]
- Kashyap, S.S.; Verma, S.; Voronin, D.; Lustigman, S.; Kulke, D.; Robertson, A.P.; Martin, R.J. Emodepside has sex-dependent immobilizing effects on adult Brugia malayi due to a differentially spliced binding pocket in the RCK1 region of the SLO-1 K channel. PLoS Pathog. 2019, 15, e1008041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maertens, J.A. History of the development of azole derivatives. Clin. Microbiol. Infect. 2004, 10 (Suppl. 1), 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrian-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, R.; Elfawal, M.A.; Wildman, S.A.; Helander, J.; Bulman, C.A.; Sakanari, J.; Rosa, B.A.; Brindley, P.J.; Janetka, J.W.; Aroian, R.V.; et al. Identification of small molecule enzyme inhibitors as broad-spectrum anthelmintics. Sci. Rep. 2019, 9, 9085. [Google Scholar] [CrossRef] [Green Version]
- Seidler, J.; McGovern, S.L.; Doman, T.N.; Shoichet, B.K. Identification and prediction of promiscuous aggregating inhibitors among known drugs. J. Med. Chem. 2003, 46, 4477–4486. [Google Scholar] [CrossRef]
- Choi, S.R.; Pradhan, A.; Hammond, N.L.; Chittiboyina, A.G.; Tekwani, B.L.; Avery, M.A. Design, synthesis, and biological evaluation of Plasmodium falciparum lactate dehydrogenase inhibitors. J. Med. Chem. 2007, 50, 3841–3850. [Google Scholar] [CrossRef]
- Mast, N.; Zheng, W.; Stout, C.D.; Pikuleva, I.A. Antifungal Azoles: Structural Insights into Undesired Tight Binding to Cholesterol-Metabolizing CYP46A1. Mol. Pharmacol. 2013, 84, 86–94. [Google Scholar] [CrossRef] [Green Version]
- Janes, J.; Young, M.E.; Chen, E.; Rogers, N.H.; Burgstaller-Muehlbacher, S.; Hughes, L.D.; Love, M.S.; Hull, M.V.; Kuhen, K.L.; Woods, A.K.; et al. The ReFRAME library as a comprehensive drug repurposing library and its application to the treatment of cryptosporidiosis. Proc. Natl. Acad. Sci. USA 2018, 115, 10750–10755. [Google Scholar] [CrossRef] [Green Version]
- Eder, J.; Hommel, U.; Cumin, F.; Martoglio, B.; Gerhartz, B. Aspartic proteases in drug discovery. Curr. Pharm. Des. 2007, 13, 271–285. [Google Scholar] [CrossRef]
- Nguyen, J.T.; Hamada, Y.; Kimura, T.; Kiso, Y. Design of potent aspartic protease inhibitors to treat various diseases. Arch. Pharm. (Weinheim) 2008, 341, 523–535. [Google Scholar] [CrossRef] [PubMed]
- WHO. The Selection and Use of Essential Medicines: Report of the WHO Expert Committee on Selection and Use of Essential Medicines, 2019 (Including the 21st WHO Model List of Essential Medicines and the 7th WHO Model List of Essential Medicines for Children); WHO: Geneva, Switzerland, 2019. [Google Scholar]
- Tort, J.; Brindley, P.J.; Knox, D.; Wolfe, K.H.; Dalton, J.P. Proteinases and associated genes of parasitic helminths. Adv. Parasitol. 1999, 43, 161–266. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liu, R.D.; Bai, S.J.; Hao, H.N.; Yue, W.W.; Xu, Y.X.Y.; Long, S.R.; Cui, J.; Wang, Z.Q. Molecular characterization of a Trichinella spiralis aspartic protease and its facilitation role in larval invasion of host intestinal epithelial cells. PLoS Negl. Trop. Dis. 2020, 14, e0008269. [Google Scholar] [CrossRef] [PubMed]
- Park, J.N.; Park, S.K.; Cho, M.K.; Park, M.K.; Kang, S.A.; Kim, D.H.; Yu, H.S. Molecular characterization of 45 kDa aspartic protease of Trichinella spiralis. Vet. Parasitol. 2012, 190, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Williamson, A.L.; Brindley, P.J.; Abbenante, G.; Datu, B.J.; Prociv, P.; Berry, C.; Girdwood, K.; Pritchard, D.I.; Fairlie, D.P.; Hotez, P.J.; et al. Hookworm aspartic protease, Na-APR-2, cleaves human hemoglobin and serum proteins in a host-specific fashion. J. Infect. Dis. 2003, 187, 484–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolodar, A.; Fischer, P.; Buttner, D.W.; Miller, D.J.; Schmetz, C.; Brattig, N.W. Onchocerca volvulus: Expression and immunolocalization of a nematode cathepsin D-like lysosomal aspartic protease. Exp. Parasitol. 2004, 107, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Hamada, Y.; Kiso, Y. New directions for protease inhibitors directed drug discovery. Biopolymers 2016, 106, 563–579. [Google Scholar] [CrossRef]
- Drag, M.; Salvesen, G.S. Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discov. 2010, 9, 690–701. [Google Scholar] [CrossRef] [Green Version]
- Monika, S.; Malgorzata, B.; Zbigniew, O. Contribution of Aspartic Proteases in Candida Virulence. Protease Inhibitors against Candida Infections. Curr. Protein Pept. Sci. 2017, 18, 1050–1062. [Google Scholar] [CrossRef]
- Castilho, V.V.S.; Goncalves, K.C.S.; Rebello, K.M.; Baptista, L.P.R.; Sangenito, L.S.; Santos, H.L.C.; Branquinha, M.H.; Santos, A.L.S.; Menna-Barreto, R.F.S.; Guimaraes, A.C.; et al. Docking simulation between HIV peptidase inhibitors and Trypanosoma cruzi aspartyl peptidase. BMC Res. Notes 2018, 11, 825. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, N.D.; Barrett, A.J.; Finn, R. Twenty years of the MEROPS database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2016, 44, D343–D350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benes, P.; Vetvicka, V.; Fusek, M. Cathepsin D-many functions of one aspartic protease. Crit. Rev. Oncol. Hematol. 2008, 68, 12–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkis, G.J.; Kurpiewski, M.R.; Ashcom, J.D.; Jen-Jacobson, L.; Jacobson, L.A. Proteases of the nematode Caenorhabditis elegans. Arch. Biochem. Biophys. 1988, 261, 80–90. [Google Scholar] [CrossRef]
- Lv, Z.; Chu, Y.; Wang, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV AIDS (Auckl.) 2015, 7, 95–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotton, J.A.; Bennuru, S.; Grote, A.; Harsha, B.; Tracey, A.; Beech, R.; Doyle, S.R.; Dunn, M.; Hotopp, J.C.; Holroyd, N.; et al. The genome of Onchocerca volvulus, agent of river blindness. Nat. Microbiol. 2016, 2, 16216. [Google Scholar] [CrossRef] [PubMed]
- Rigobello, M.P.; Scutari, G.; Boscolo, R.; Bindoli, A. Induction of mitochondrial permeability transition by auranofin, a gold(I)-phosphine derivative. Br. J. Pharmacol. 2002, 136, 1162–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Njouendou, A.J.; Ritter, M.; Ndongmo, W.P.C.; Kien, C.A.; Narcisse, G.T.V.; Fombad, F.F.; Tayong, D.B.; Pfarr, K.; Layland, L.E.; Hoerauf, A.; et al. Successful long-term maintenance of Mansonella perstans in an in vitro culture system. Parasites Vectors 2017, 10, 563. [Google Scholar] [CrossRef] [Green Version]
- Verma, M.; Pathak, M.; Shahab, M.; Singh, K.; Mitra, K.; Misra-Bhattacharya, S. Moxidectin causes adult worm mortality of human lymphatic filarial parasite Brugia malayi in rodent models. Folia Parasitol. (Praha) 2014, 61, 561–570. [Google Scholar] [CrossRef]
- Verma, S.; Kashyap, S.S.; Robertson, A.P.; Martin, R.J. Functional genomics in Brugia malayi reveal diverse muscle nAChRs and differences between cholinergic anthelmintics. Proc. Natl. Acad. Sci. USA 2017, 114, 5539–5544. [Google Scholar] [CrossRef] [Green Version]
- Partridge, F.A.; Forman, R.; Bataille, C.J.R.; Wynne, G.M.; Nick, M.; Russell, A.J.; Else, K.J.; Sattelle, D.B. Anthelmintic drug discovery: Target identification, screening methods and the role of open science. Beilstein J. Org. Chem. 2020, 16, 1203–1224. [Google Scholar] [CrossRef]
- Marcellino, C. GitHub Repository. WormAssay. Available online: https://github.com/chrismarcellino/wormassay. (accessed on 23 September 2020).
- Bulman, C.A.; Bidlow, C.M.; Lustigman, S.; Cho-Ngwa, F.; Williams, D.; Rascon, A.A., Jr.; Tricoche, N.; Samje, M.; Bell, A.; Suzuki, B.; et al. Repurposing auranofin as a lead candidate for treatment of lymphatic filariasis and onchocerciasis. PLoS Negl. Trop. Dis. 2015, 9, e0003534. [Google Scholar] [CrossRef] [PubMed]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, T.W.; Arnaboldi, V.; Cain, S.; Chan, J.; Chen, W.J.; Cho, J.; Davis, P.; Gao, S.; Grove, C.A.; Kishore, R.; et al. WormBase: A modern Model Organism Information Resource. Nucleic Acids Res. 2020, 48, D762–D767. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Charisi, A.; Cheng, L.C.; Jiang, T.; Girke, T. ChemmineR: A compound mining framework for R. Bioinformatics 2008, 24, 1733–1734. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Common Name | Compound Class | Compound Subclass | Literature Targets (Not All are Listed) | Brugia pahangi (Adult Female) | ||
|---|---|---|---|---|---|---|
| % Motility Inhibition (10 μM) | IC50 (μM) | |||||
| Group A | Econazole | Imidazole | Benzyl ether | Sterol 14-demethylase/K+ VGIC [72] | 100 | 4.0 |
| Miconazole | Imidazole | Benzyl ether | Sterol 14-demethylase/K+ VGIC [72] | 96 | 3.3 | |
| Sulconazole | Imidazole | Thiobenzyl ether | Sterol 14-demethylase [73] | 98 | 4.6 | |
| Sertaconazole | Imidazole | Benzothiophene | Sterol 14-demethylase [74] | 100 | - | |
| Clotrimazole | Imidazole | Triphenylmethane | Sterol 14-demethylase/K+ VGIC [75] | 85 | 5.5 | |
| Itraconazole | Triazole | Piperazinyl | XIAP/K+ VGIC [76,77] | 80 | - | |
| Group B | Suloctidil | Amino alcohol | Phenyl thioether | K+ VGIC/thyroid receptor [52,53] | 99 | 3.7 |
| Pimozide | Benzimidazolone | Piperidine | DRD2/DRD3 (GPCRs)/KCNH2/Calmodulin [54,55,56] | 96 | 2.5 | |
| Butamben | Benzoic ester | Aniline | VGICs [57] | 66 | - | |
| Clemizole | Benzimidazole | Pyrrolidine | HRH1/K+ VGIC/NR ROR-gamma [58,59] | 78 | 6.5 | |
| Proroxan | Phenyl ketone | Phenyl pyrrolidine | Adrenergic receptors [60] | 65 | 8.5 | |
| Saccharin | Benzoisothiazolone | Sulfonyl amide | carbonic anhydrase [61] | 57 | - | |
| Ritonavir | Amino alcohol | Peptidomimetic | HIV protease/CYP3A/SLC47 [62,63,64] | 52 | - | |
| Nifenazone | Dihydropyrazole | Nicotinamide | - | 50 | - | |
| Levonordefrin | Amino alcohol | Phenol | ADRA2/HADH2/TF HIF1A/APE1 [65,66] | 54 | - | |
| Tafenoquine | Quinoline | Alkyl amine | cytochrome c reductase [67] | 63 | 14.1 | |
| Primaquine | Quinoline | Alkyl amine | quinone reductase [68,69] | 51 | 5.1 | |
| Apomorphine | Aporphine | Tetrahydroisoquinoline | Dopamine/serotonin/adrenergic receptor agonist [70,71] | 53 | - | |
| Drug Common Name | Day 3 Adult Female B. pahangi | Day 7 Adult Female O. ochengi | Day 5 Adult Male O. ochengi | Day 28 O. volvulus L5 | Day 5 Loa loa mf | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| % Inhibition of Motility (10 μM) | IC50 (μM) | % Inhibition of Viability (10 μM) | IC50 (μM) | % Inhibition of Motility (10 μM) | IC50 (μM) | % Inhibition of Motility (10 μM) | IC50 (μM) | % Inhibition of Motility (10 μM) | IC50 (μM) | ||
| Group A | Isoconazole | 100 | 3.8 | 57 | 30.8 | 100 | 0.6 | 96 | 2.4 | 33.3 | 10 |
| Fenticonazole | 100 | 1.2 | 32 | 94.7 | 75 | 10.6 | 83 | 4.7 | 50 | 10 | |
| Sertaconazole | 100 | - | 21 | - | 31 | - | - | - | - | - | |
| Econazole | 100 | 4.0 | 22 | - | 26 | - | - | - | - | - | |
| Tioconazole | 99 | 3.2 | 14 | - | 100 | - | - | - | - | - | |
| Sulconazole | 98 | 4.6 | 26 | - | 99 | - | - | - | - | - | |
| Miconazole | 96 | 3.3 | 0 | - | 100 | - | - | - | - | - | |
| Clotrimazole | 85 | 5.5 | 8 | - | 100 | - | - | - | - | - | |
| Posaconazole | 82 | 0.1 | 38 | >100 | 79 | 6.1 | 57 * | - | - | - | |
| Itraconazole | 80 | - | 13 | - | 21 | - | - | - | - | - | |
| Group B | Suloctidil | 99 | 3.7 | 87 | 4.1 | 100 | 5.5 | 100 ** | - | 100 | 4.1 |
| Pimozide | 96 | 2.5 | 13 | - | 96 | - | - | - | - | - | |
| Primaquine | 51 | 5.1 | 100 | 1.3 | 100 | 0.4 | 100 ** | - | 50 | 18.5 | |
| Aspartyl Protease Inhibitors (APIs) | % Inhibition of Motility (30 µM) | % Inhibition of Molting (L3 to L4) | ||||
|---|---|---|---|---|---|---|
| B. pahangi | O. ochengi | O. volvulus | O. volvulus (day 6) | |||
| Female (day 6) | Female (day 7) ** | Male (day 5) | L4 (day 7) | 3 μM | 10 μM | |
| Nelfinavir | 99 | 100 | 100 | 73 | 50.2 | 45.5 |
| Lopinavir | 98 | 0 | 88 | 56 | 0 | 74.1 |
| Ritonavir | 52 * | 63 | 100 | 0 | 0 | 0 |
| Pepstatin A | 0 | 50 | 100 | 0 | 0 | 40 |
| Darunavir | 0 | 30 | ND | 4.4 | 43.9 | 46.7 |
| Aliskiren | 0 | 6 | ND | 0 | 30.4 | 51.7 |
| Amprenavir | 24 | 0 | 17 | 8 | 0 | 43.5 |
| Atazanavir | 11 | 42 | ND | 30 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tyagi, R.; Bulman, C.A.; Cho-Ngwa, F.; Fischer, C.; Marcellino, C.; Arkin, M.R.; McKerrow, J.H.; McNamara, C.W.; Mahoney, M.; Tricoche, N.; et al. An Integrated Approach to Identify New Anti-Filarial Leads to Treat River Blindness, a Neglected Tropical Disease. Pathogens 2021, 10, 71. https://doi.org/10.3390/pathogens10010071
Tyagi R, Bulman CA, Cho-Ngwa F, Fischer C, Marcellino C, Arkin MR, McKerrow JH, McNamara CW, Mahoney M, Tricoche N, et al. An Integrated Approach to Identify New Anti-Filarial Leads to Treat River Blindness, a Neglected Tropical Disease. Pathogens. 2021; 10(1):71. https://doi.org/10.3390/pathogens10010071
Chicago/Turabian StyleTyagi, Rahul, Christina A. Bulman, Fidelis Cho-Ngwa, Chelsea Fischer, Chris Marcellino, Michelle R. Arkin, James H. McKerrow, Case W. McNamara, Matthew Mahoney, Nancy Tricoche, and et al. 2021. "An Integrated Approach to Identify New Anti-Filarial Leads to Treat River Blindness, a Neglected Tropical Disease" Pathogens 10, no. 1: 71. https://doi.org/10.3390/pathogens10010071