A Review of Multi-Scale Computational Modeling Tools for Predicting Structures and Properties of Multi-Principal Element Alloys


Abstract
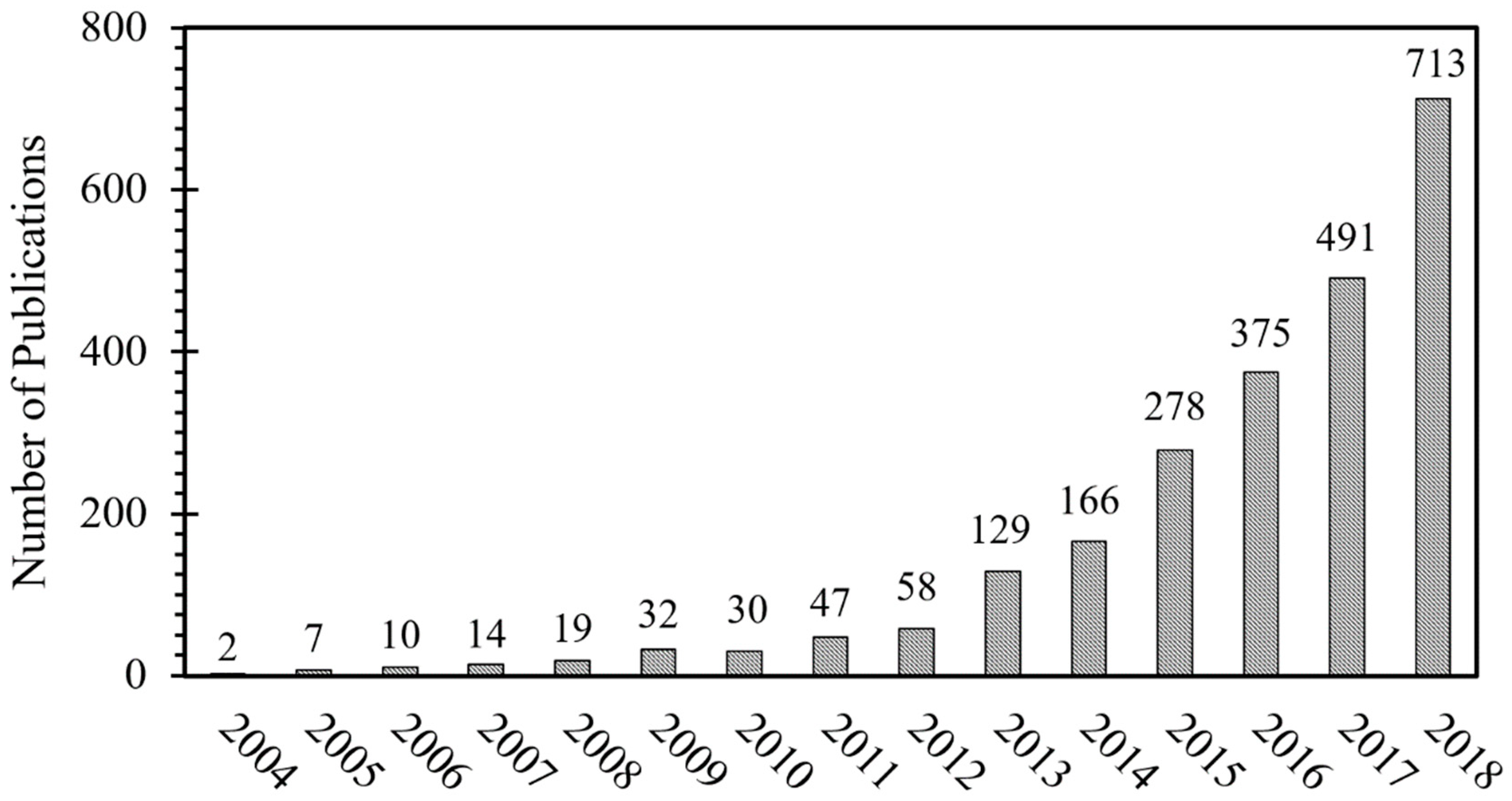
:1. Introduction
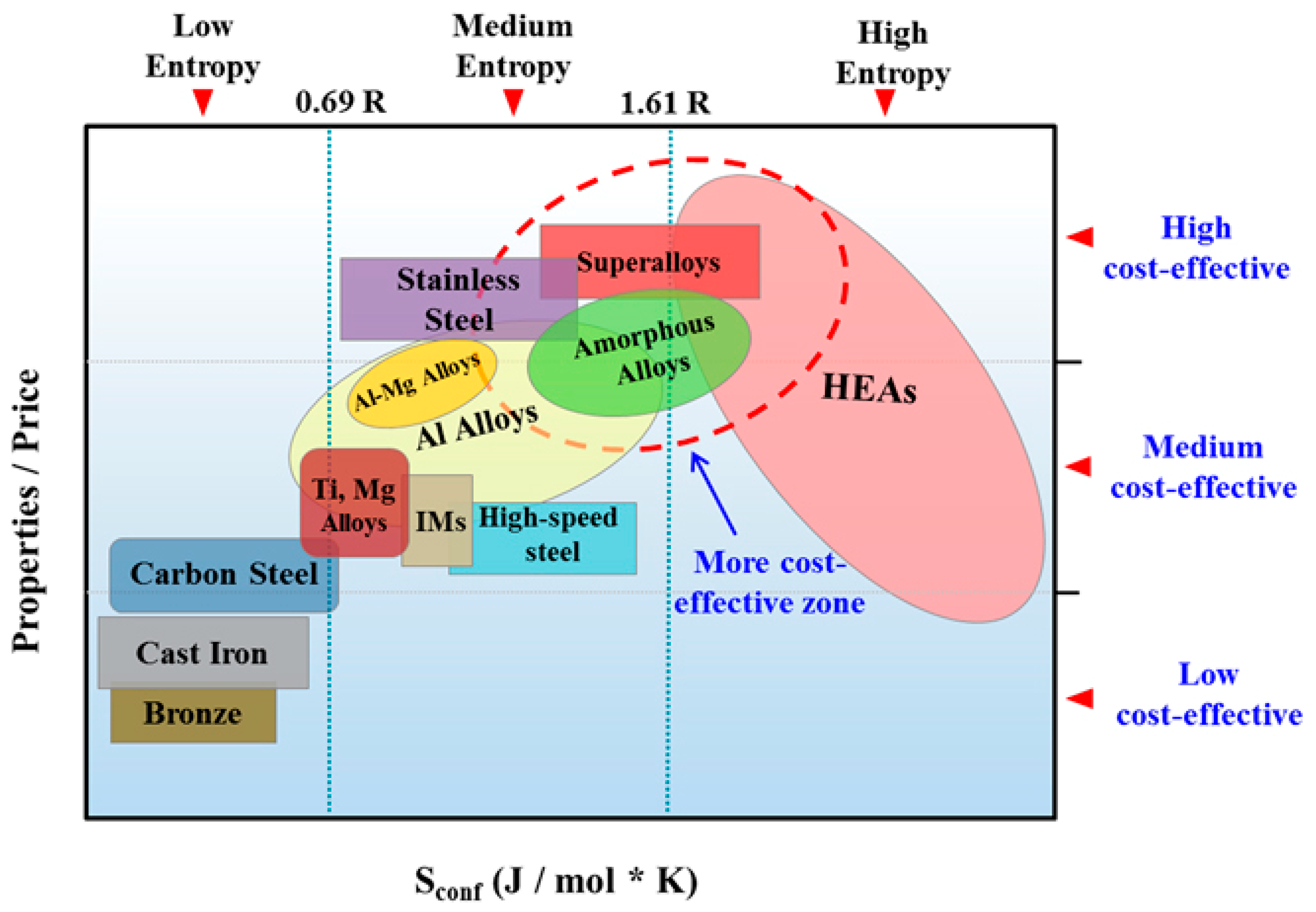
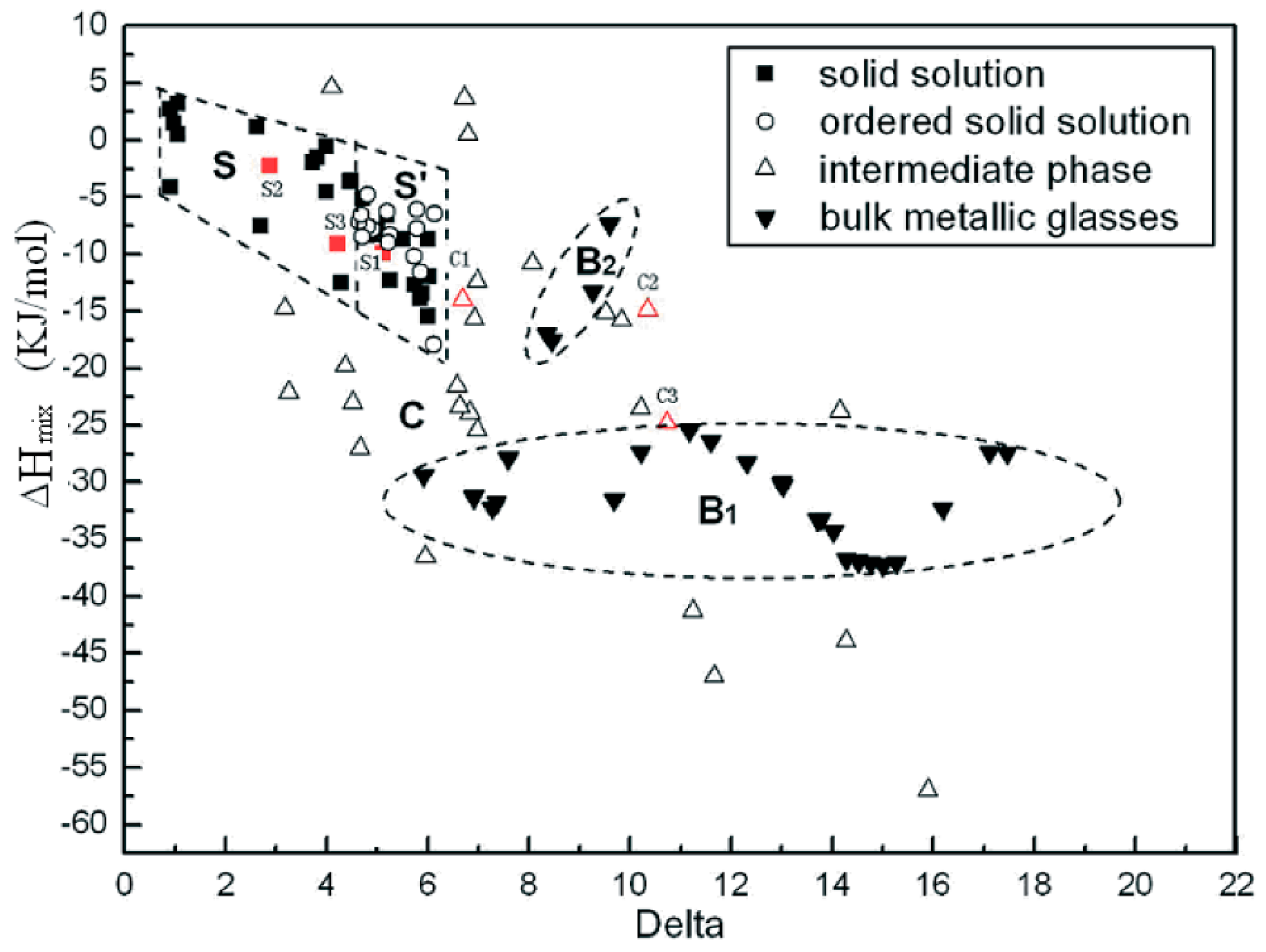
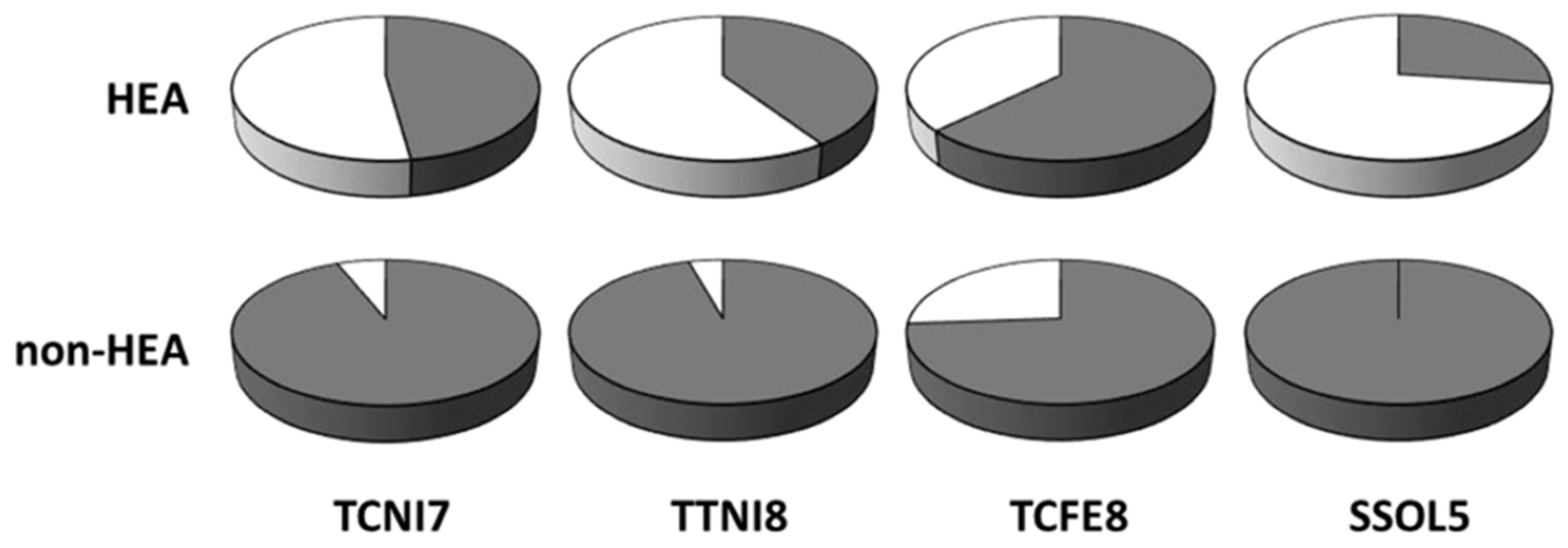
2. Phase Equilibria and Crystal Structures of MPE Alloys
3. Properties of MPE Alloys
3.1. Mechanical Properties
3.1.1. Elastic Properties
3.1.2. Plastic Deformation
3.1.3. Solute Strengthening
3.2. Thermo-Chemical Properties
3.3. Magnetic Properties
4. Summary
Author Contributions
Conflicts of Interest
References
- Miracle, D.; Senkov, O. A critical review of high entropy alloys and related concepts. Acta Mater. 2017, 122, 448–511. [Google Scholar] [CrossRef] [Green Version]
- Murty, B.S.; Yeh, J.-W.; Ranganathan, S. High-Entropy Alloys; Butterworth-Heinemann: Oxford, UK, 2014. [Google Scholar]
- Zhang, C.; Zhang, F.; Chen, S.; Cao, W. Computational thermodynamics aided high-entropy alloy design. JOM 2012, 64, 839–845. [Google Scholar] [CrossRef]
- Yeh, J.-W. Alloy Design Strategies and Future Trends in High-Entropy Alloys. JOM 2013, 65, 1759–1771. [Google Scholar] [CrossRef]
- Tsai, K.-Y.; Tsai, M.-H.; Yeh, J.-W. Sluggish diffusion in Co–Cr–Fe–Mn–Ni high-entropy alloys. Acta Mater. 2013, 61, 4887–4897. [Google Scholar] [CrossRef]
- Wu, Z.; Parish, C.; Bei, H. Nano-twin mediated plasticity in carbon-containing FeNiCoCrMn high entropy alloys. J. Alloy. Compd. 2015, 647, 815–822. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Baker, I. Interstitial strengthening of a fcc FeNiMnAlCr high entropy alloy. Mater. Lett. 2016, 180, 153–156. [Google Scholar] [CrossRef]
- Li, Z.; Tasan, C.C.; Springer, H.; Gault, B.; Raabe, D. Interstitial atoms enable joint twinning and transformation induced plasticity in strong and ductile high-entropy alloys. Sci. Rep. 2017, 7, 40704. [Google Scholar] [CrossRef] [Green Version]
- Laurent-Brocq, M.; Sauvage, X.; Akhatova, A.; Perrière, L.; Leroy, E.; Champion, Y. Precipitation and Hardness of Carbonitrides in a CrMnFeCoNi High Entropy Alloy. Adv. Eng. Mater. 2017, 19. [Google Scholar] [CrossRef]
- Beyramali Kivy, M.; Kriewall, C.S.; Zaeem, M.A. Formation of chromium-iron carbide by carbon diffusion in Al X CoCrFeNiCu high-entropy alloys. Mater. Res. Lett. 2018, 6, 321–326. [Google Scholar] [CrossRef]
- Fu, X.; Schuh, C.; Olivetti, E. Materials selection considerations for high entropy alloys. Scr. Mater. 2017, 138, 145–150. [Google Scholar] [CrossRef]
- Yeh, J.W.; Chen, S.K.; Lin, S.J.; Gan, J.Y.; Chin, T.S.; Shun, T.T.; Tsau, C.H.; Chang, S.Y. Nanostructured high-entropy alloys with multiple principal elements: Novel alloy design concepts and outcomes. Adv. Eng. Mater. 2004, 6, 299–303. [Google Scholar] [CrossRef]
- Ye, Y.; Wang, Q.; Zhao, Y.; He, Q.; Lu, J.; Yang, Y. Elemental segregation in solid-solution high-entropy alloys: Experiments and modeling. J. Alloy. Compd. 2016, 681, 167–174. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, X.; Ma, J.; Lu, Z.; Zhao, Y. Compositional gradient films constructed by sputtering in a multicomponent Ti–Al–(Cr, Fe, Ni) system. J. Mater. Res. 2018, 33, 3330–3338. [Google Scholar] [CrossRef]
- Sheng, G.; Liu, C.T. Phase stability in high entropy alloys: Formation of solid-solution phase or amorphous phase. Prog. Natl. Sci. Mater. Int. 2011, 21, 433–446. [Google Scholar] [Green Version]
- Pickering, E.; Jones, N.G. High-entropy alloys: A critical assessment of their founding principles and future prospects. Int. Mater. Rev. 2016, 61, 183–202. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, Y.J.; Lin, J.P.; Chen, G.L.; Liaw, P.K. Solid-Solution Phase Formation Rules for Multi-component Alloys. Adv. Eng. Mater. 2008, 10, 534–538. [Google Scholar] [CrossRef]
- Mizutani, U. The Hume-Rothery Rules for Structurally Complex Alloy Phases, in Surface Properties and Engineering of Complex Intermetallics; World Scientific: Singapore, 2010; pp. 323–399. [Google Scholar]
- Guo, S.; Ng, C.; Lu, J.; Liu, C. Effect of valence electron concentration on stability of fcc or bcc phase in high entropy alloys. J. Appl. Phys. 2011, 109, 103505. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.-H.; Tsai, K.-Y.; Tsai, C.-W.; Lee, C.; Juan, C.-C.; Yeh, J.-W. Criterion for sigma phase formation in Cr-and V-containing high-entropy alloys. Mater. Res. Lett. 2013, 1, 207–212. [Google Scholar] [CrossRef]
- Tsai, M.-H.; Yeh, J.-W. High-entropy alloys: A critical review. Mater. Res. Lett. 2014, 2, 107–123. [Google Scholar] [CrossRef]
- Morral, J.; Chen, S.-L. High Entropy Alloys, Miscibility Gaps and the Rose Geometry. J. Phase Equilib. Diffus. 2017, 38, 319–331. [Google Scholar] [CrossRef]
- Kaufman, L.; Bernstein, H. Computer Calculation of Phase Diagrams. With Special Reference to Refractory Metals; Academic Press: Cambridge, MA, USA, 1970. [Google Scholar]
- Kroupa, A. Modelling of phase diagrams and thermodynamic properties using Calphad method—Development of thermodynamic databases. Comput. Mater. Sci. 2013, 66, 3–13. [Google Scholar] [CrossRef]
- Kattner, U.R. The thermodynamic modeling of multicomponent phase equilibria. JOM 1997, 49, 14–19. [Google Scholar] [CrossRef]
- Bale, C.; Bélisle, E.; Chartrand, P.; Decterov, S.; Eriksson, G.; Hack, K.; Jung, I.-H.; Kang, Y.-B.; Melançon, J.; Pelton, A. FactSage thermochemical software and databases—Recent developments. Calphad 2009, 33, 295–311. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J. First principles study of structural and thermodynamic properties of zirconia. Mater. Today Proc. 2014, 1, 44–54. [Google Scholar] [CrossRef]
- Chen, H.-L.; Mao, H.; Chen, Q. Database development and Calphad calculations for high entropy alloys: Challenges, strategies, and tips. Mater. Chem. Phys. 2018, 210, 279–290. [Google Scholar] [CrossRef]
- CompuTherm, L. Pandat 8.0-Phase Diagram Calculation Software for Multi-Component Systems; CompuTherm LLC: Madison, WI, USA, 2008; Volume 53719. [Google Scholar]
- Idury, K.S.; Murty, B.; Bhatt, J. Thermodynamic modeling and composition design for the formation of Zr–Ti–Cu–Ni–Al high entropy bulk metallic glasses. Intermetallics 2015, 65, 42–50. [Google Scholar] [CrossRef]
- Idury, K.S.; Murty, B.; Bhatt, J. Identifying non-equiatomic high entropy bulk metallic glass formers through thermodynamic approach: A theoretical perspective. J. Non-Cryst. Solids 2016, 450, 164–173. [Google Scholar] [CrossRef]
- Tang, Z.; Gao, M.C.; Diao, H.; Yang, T.; Liu, J.; Zuo, T.; Zhang, Y.; Lu, Z.; Cheng, Y.; Zhang, Y. Aluminum alloying effects on lattice types, microstructures, and mechanical behavior of high-entropy alloys systems. JOM 2013, 65, 1848–1858. [Google Scholar] [CrossRef]
- Sonkusare, R.; Janani, P.D.; Gurao, N.; Sarkar, S.; Sen, S.; Pradeep, K.; Biswas, K. Phase equilibria in equiatomic CoCuFeMnNi high entropy alloy. Mater. Chem. Phys. 2018, 210, 269–278. [Google Scholar] [CrossRef]
- Saal, J.E.; Berglund, I.S.; Sebastian, J.T.; Liaw, P.K.; Olson, G.B. Equilibrium high entropy alloy phase stability from experiments and thermodynamic modeling. Scr. Mater. 2018, 146, 5–8. [Google Scholar] [CrossRef]
- Yao, H.; Qiao, J.; Gao, M.; Hawk, J.; Ma, S.; Zhou, H.; Zhang, Y. NbTaV-(Ti, W) refractory high-entropy alloys: Experiments and modeling. Mater. Sci. Eng. A 2016, 674, 203–211. [Google Scholar] [CrossRef]
- Feng, R.; Gao, M.C.; Lee, C.; Mathes, M.; Zuo, T.; Chen, S.; Hawk, J.A.; Zhang, Y.; Liaw, P.K. Design of light-weight high-entropy alloys. Entropy 2016, 18, 333. [Google Scholar] [CrossRef]
- Stepanov, N.; Yurchenko, N.Y.; Skibin, D.; Tikhonovsky, M.; Salishchev, G. Structure and mechanical properties of the AlCrxNbTiV (x = 0, 0.5, 1, 1.5) high entropy alloys. J. Alloy. Compd. 2015, 652, 266–280. [Google Scholar] [CrossRef]
- Gwalani, B.; Gorsse, S.; Choudhuri, D.; Styles, M.; Zheng, Y.; Mishra, R.S.; Banerjee, R. Modifying transformation pathways in high entropy alloys or complex concentrated alloys via thermo-mechanical processing. Acta Mater. 2018, 153, 169–185. [Google Scholar] [CrossRef]
- Abu-Odeh, A.; Galvan, E.; Kirk, T.; Mao, H.; Chen, Q.; Mason, P.; Malak, R.; Arróyave, R. Efficient exploration of the High Entropy Alloy composition-phase space. Acta Mater. 2018. [Google Scholar] [CrossRef]
- Tancret, F.; Toda-Caraballo, I.; Menou, E.; Díaz-Del, P.E.J.R. Designing high entropy alloys employing thermodynamics and Gaussian process statistical analysis. Mater. Des. 2017, 115, 486–497. [Google Scholar] [CrossRef]
- Gorsse, S.; Tancret, F. Current and emerging practices of CALPHAD toward the development of high entropy alloys and complex concentrated alloys. J. Mater. Res. 2018, 33, 2899–2923. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, S.; Gao, M.C.; Zhang, C.; Zhang, T.; Yang, H.; Wang, Z.; Qiao, J. Tribological properties of AlCrCuFeNi2 high-entropy alloy in different conditions. Metall. Mater. Trans. A 2016, 47, 3312–3321. [Google Scholar] [CrossRef]
- Haase, C.; Tang, F.; Wilms, M.B.; Weisheit, A.; Hallstedt, B. Combining thermodynamic modeling and 3D printing of elemental powder blends for high-throughput investigation of high-entropy alloys–Towards rapid alloy screening and design. Mater. Sci. Eng. A 2017, 688, 180–189. [Google Scholar] [CrossRef]
- Arslan, H.; Dogan, A. Thermodynamic investigations on the component dependences of high-entropy alloys. Russ. J. Phys. Chem. A 2016, 90, 2339–2345. [Google Scholar] [CrossRef]
- Eshed, E.; Larianovsky, N.; Kovalevsky, A.; Popov, V., Jr.; Gorbachev, I.; Popov, V.; Katz-Demyanetz, A. Microstructural Evolution and Phase Formation in 2nd-Generation Refractory-Based High Entropy Alloys. Materials 2018, 11, 175. [Google Scholar] [CrossRef] [PubMed]
- Beyramali Kivy, M.; Zaeem, M.A.; Lekakh, S. Investigating phase formations in cast AlFeCoNiCu high entropy alloys by combination of computational modeling and experiments. Mater. Des. 2017, 127, 224–232. [Google Scholar] [CrossRef]
- Toda-Caraballo, I.; Wróbel, J.; Nguyen-Manh, D.; Perez, P.; Rivera-Díaz-del-Castillo, P. Simulation and modeling in high entropy alloys. JOM 2017, 69, 2137–2149. [Google Scholar] [CrossRef]
- Kresse, G.; Marsman, O.; Furthmuller, J. VASP the Guide. Available online: http://cms.mpi.univie.ac.at/vasp/vasp.pdf (accessed on 24 April 2016).
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Gonze, X.; Beuken, J.-M.; Caracas, R.; Detraux, F.; Fuchs, M.; Rignanese, G.-M.; Sindic, L.; Verstraete, M.; Zerah, G.; Jollet, F. First-principles computation of material properties: The ABINIT software project. Comput. Mater. Sci. 2002, 25, 478–492. [Google Scholar] [CrossRef]
- Segall, M.; Lindan, P.J.; Probert, M.A.; Pickard, C.J.; Hasnip, P.J.; Clark, S.; Payne, M. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717. [Google Scholar] [CrossRef]
- Ma, D.; Grabowski, B.; Körmann, F.; Neugebauer, J.; Raabe, D. Ab initio thermodynamics of the CoCrFeMnNi high entropy alloy: Importance of entropy contributions beyond the configurational one. Acta Mater. 2015, 100, 90–97. [Google Scholar] [CrossRef]
- Niu, C.; Zaddach, A.; Koch, C.; Irving, D. First principles exploration of near-equiatomic NiFeCrCo high entropy alloys. J. Alloy. Compd. 2016, 672, 510–520. [Google Scholar] [CrossRef] [Green Version]
- Yalamanchili, K.; Wang, F.; Schramm, I.; Andersson, J.; Jöesaar, M.J.; Tasnadi, F.; Muecklich, F.; Ghafoor, N.; Odén, M. Exploring the high entropy alloy concept in (AlTiVNbCr) N. Thin Solid Films 2017, 636, 346–352. [Google Scholar] [CrossRef]
- Jiang, C.; Uberuaga, B.P. Efficient ab initio modeling of random multicomponent alloys. Phys. Rev. Lett. 2016, 116, 105501. [Google Scholar] [CrossRef]
- Tian, F.; Varga, L.K.; Vitos, L. Predicting single phase CrMoWX high entropy alloys from empirical relations in combination with first-principles calculations. Intermetallics 2017, 83, 9–16. [Google Scholar] [CrossRef]
- Li, Z.; Körmann, F.; Grabowski, B.; Neugebauer, J.; Raabe, D. Ab initio assisted design of quinary dual-phase high-entropy alloys with transformation-induced plasticity. Acta Mater. 2017, 136, 262–270. [Google Scholar] [CrossRef]
- Heidelmann, M.; Feuerbacher, M.; Ma, D.; Grabowski, B. Structural anomaly in the high-entropy alloy ZrNbTiTaHf. Intermetallics 2016, 68, 11–15. [Google Scholar] [CrossRef]
- Mu, Y.; Liu, H.; Liu, Y.; Zhang, X.; Jiang, Y.; Dong, T. An ab initio and experimental studies of the structure, mechanical parameters and state density on the refractory high-entropy alloy systems. J. Alloy. Compd. 2017, 714, 668–680. [Google Scholar] [CrossRef]
- Zhang, F.; Zhao, S.; Jin, K.; Bei, H.; Popov, D.; Park, C.; Neuefeind, J.C.; Weber, W.J.; Zhang, Y. Pressure-induced fcc to hcp phase transition in Ni-based high entropy solid solution alloys. Appl. Phys. Lett. 2017, 110, 011902. [Google Scholar] [CrossRef]
- Tian, F.; Delczeg, L.; Chen, N.; Varga, L.K.; Shen, J.; Vitos, L. Structural stability of NiCoFeCrAl x high-entropy alloy from ab initio theory. Phys. Rev. B 2013, 88, 085128. [Google Scholar] [CrossRef]
- Middleburgh, S.; King, D.; Lumpkin, G.; Cortie, M.; Edwards, L. Segregation and migration of species in the CrCoFeNi high entropy alloy. J. Alloy. Compd. 2014, 599, 179–182. [Google Scholar] [CrossRef]
- Takaki, T.; Ohno, M.; Shibuta, Y.; Sakane, S.; Shimokawabe, T.; Aoki, T. Two-dimensional phase-field study of competitive grain growth during directional solidification of polycrystalline binary alloy. J. Cryst. Growth 2016, 442, 14–24. [Google Scholar] [CrossRef]
- Leong, Z.; Wróbel, J.S.; Dudarev, S.L.; Goodall, R.; Todd, I.; Nguyen-Manh, D. The effect of electronic structure on the phases present in high entropy alloys. Sci. Rep. 2017, 7, 39803. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.; Rodriguez, G.; Bozzolo, G.; Mosca, H. Melting temperature of CoCrFeNiMn high-entropy alloys. Comput. Mater. Sci. 2018, 148, 69–75. [Google Scholar] [CrossRef]
- Zhang, Y.; Stocks, G.M.; Jin, K.; Lu, C.; Bei, H.; Sales, B.C.; Wang, L.; Béland, L.K.; Stoller, R.E.; Samolyuk, G.D. Influence of chemical disorder on energy dissipation and defect evolution in concentrated solid solution alloys. Nat. Commun. 2015, 6, 8736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pielnhofer, F.; Schöneich, M.; Lorenz, T.; Yan, W.; Nilges, T.; Weihrich, R.; Schmidt, P. A Rational Approach to IrPTe–DFT and CalPhaD Studies on Phase Stability, Formation, and Structure of IrPTe. Z. Anorg. Allg. Chem. 2015, 641, 1099–1105. [Google Scholar] [CrossRef]
- Mathieu, R.; Dupin, N.; Crivello, J.-C.; Yaqoob, K.; Breidi, A.; Fiorani, J.-M.; David, N.; Joubert, J.-M. CALPHAD description of the Mo–Re system focused on the sigma phase modeling. Calphad 2013, 43, 18–31. [Google Scholar] [CrossRef] [Green Version]
- Bigdeli, S. Developing the Third Generation of Calphad Databases: What Can ab-Initio Contribute? KTH Royal Institute of Technology: Stockholm, Sweden, 2017. [Google Scholar]
- Ikeda, Y.; Grabowski, B.; Körmann, F. Ab initio phase stabilities and mechanical properties of multicomponent alloys: A comprehensive review for high entropy alloys and compositionally complex alloys. Mater. Charact. 2018, 147, 464–511. [Google Scholar] [CrossRef]
- Yao, Q.; Shang, S.-L.; Wang, K.; Liu, F.; Wang, Y.; Wang, Q.; Lu, T.; Liu, Z.-K. Phase stability, elastic, and thermodynamic properties of the L1 2 (Co, Ni) 3 (Al, Mo, Nb) phase from first-principles calculations. J. Mater. Res. 2017, 32, 2100–2108. [Google Scholar] [CrossRef]
- Gao, M.C.; Zhang, B.; Guo, S.; Qiao, J.; Hawk, J. High-entropy alloys in hexagonal close-packed structure. Metall. Mater. Trans. A 2016, 47, 3322–3332. [Google Scholar] [CrossRef]
- Gao, M.C.; Zhang, B.; Yang, S.; Guo, S. Senary refractory high-entropy alloy HfNbTaTiVZr. Metall. Mater. Trans. A 2016, 47, 3333–3345. [Google Scholar] [CrossRef]
- Choi, W.-M.; Jo, Y.H.; Sohn, S.S.; Lee, S.; Lee, B.-J. Understanding the physical metallurgy of the CoCrFeMnNi high-entropy alloy: An atomistic simulation study. npj Comput. Mater. 2018, 4, 1. [Google Scholar] [CrossRef]
- Sharma, A.; Deshmukh, S.A.; Liaw, P.K.; Balasubramanian, G. Crystallization kinetics in AlxCrCoFeNi (0 ≤ x ≤ 40) high-entropy alloys. Scr. Mater. 2017, 141, 54–57. [Google Scholar] [CrossRef]
- Mooney, C.Z. Monte Carlo Simulation; Sage Publications: Thousand Oaks, CA, USA, 1997; Volume 116. [Google Scholar]
- Anzorena, M.S.; Bertolo, A.; Gagetti, L.; Kreiner, A.; Mosca, H.; Bozzolo, G.; del Grosso, M. Characterization and modeling of a MoTaVWZr high entropy alloy. Mater. Des. 2016, 111, 382–388. [Google Scholar] [CrossRef]
- Feng, W.Q.; Zheng, S.M.; Qi, Y.; Wang, S.Q. Periodic Maximum Entropy Random Structure Models for High-Entropy Alloys. Mater. Sci. Forum 2017, 898, 611–621. [Google Scholar] [CrossRef]
- Widom, M. Modeling the structure and thermodynamics of high-entropy alloys. J. Mater. Res. 2018, 33, 2881–2898. [Google Scholar] [CrossRef]
- Wang, Z.; Fang, Q.; Li, J.; Liu, B.; Liu, Y. Effect of lattice distortion on solid solution strengthening of BCC high-entropy alloys. J. Mater. Sci. Technol. 2018, 34, 349–354. [Google Scholar] [CrossRef]
- del Grosso, M.; Bozzolo, G.; Mosca, H. Modeling of high entropy alloys of refractory elements. Phys. B Condens. Matter 2012, 407, 3285–3287. [Google Scholar] [CrossRef]
- Toda-Caraballo, I.; Rivera-Díaz-del-Castillo, P.E. Modelling and design of magnesium and high entropy alloys through combining statistical and physical models. JOM 2015, 67, 108–117. [Google Scholar] [CrossRef]
- Kucza, W.; Dąbrowa, J.; Cieślak, G.; Berent, K.; Kulik, T.; Danielewski, M. Studies of “sluggish diffusion” effect in Co-Cr-Fe-Mn-Ni, Co-Cr-Fe-Ni and Co-Fe-Mn-Ni high entropy alloys; determination of tracer diffusivities by combinatorial approach. J. Alloy. Compd. 2018, 731, 920–928. [Google Scholar] [CrossRef]
- Fernández-Caballero, A.; Fedorov, M.; Wróbel, J.S.; Mummery, P.M.; Nguyen-Manh, D. Configurational Entropy in Multicomponent Alloys: Matrix Formulation from Ab Initio Based Hamiltonian and Application to the FCC Cr-Fe-Mn-Ni System. Entropy 2019, 21, 68. [Google Scholar] [CrossRef]
- Fernandez-Caballero, A.; Wróbel, J.; Mummery, P.; Nguyen-Manh, D. Short-range order in high entropy alloys: Theoretical formulation and application to Mo-Nb-Ta-VW system. J. Phase Equilib. Diffus. 2017, 38, 391–403. [Google Scholar] [CrossRef]
- Gludovatz, B.; Hohenwarter, A.; Catoor, D.; Chang, E.H.; George, E.P.; Ritchie, R.O. A fracture-resistant high-entropy alloy for cryogenic applications. Science 2014, 345, 1153–1158. [Google Scholar] [CrossRef] [Green Version]
- Andersen, O.; Jepsen, O.; Krier, G. Exact Muffin-Tin Orbital Theory. In Lectures on Methods of Electronic Structure Calculations; World Scientific: Singapore, 1994; pp. 63–124. [Google Scholar]
- Vitos, L.; Skriver, H.L.; Johansson, B.; Kollár, J. Application of the exact muffin-tin orbitals theory: The spherical cell approximation. Comput. Mater. Sci. 2000, 18, 24–38. [Google Scholar] [CrossRef]
- Tian, F.; Varga, L.K.; Shen, J.; Vitos, L. Calculating elastic constants in high-entropy alloys using the coherent potential approximation: Current issues and errors. Comput. Mater. Sci. 2016, 111, 350–358. [Google Scholar] [CrossRef]
- Tian, F.; Varga, L.K.; Chen, N.; Shen, J.; Vitos, L. Ab initio design of elastically isotropic TiZrNbMoVx high-entropy alloys. J. Alloy. Compd. 2014, 599, 19–25. [Google Scholar] [CrossRef]
- Li, X.; Irving, D.L.; Vitos, L. First-principles investigation of the micromechanical properties of fcc-hcp polymorphic high-entropy alloys. Sci. Rep. 2018, 8, 11196. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Ni, X.; Tian, F.; Varga, L.K.; Vitos, L. Ab initio study of AlxMoNbTiV high-entropy alloys. J. Phys. Condens. Matter 2015, 27, 075401. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tian, F.; Schönecker, S.; Zhao, J.; Vitos, L. Ab initio-predicted micro-mechanical performance of refractory high-entropy alloys. Sci. Rep. 2015, 5, 12334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Schönecker, S.; Li, W.; Varga, L.K.; Irving, D.L.; Vitos, L. Tensile and shear loading of four fcc high-entropy alloys: A first-principles study. Phys. Rev. B 2018, 97, 094102. [Google Scholar] [CrossRef]
- Ge, H.; Song, H.; Shen, J.; Tian, F. Effect of alloying on the thermal-elastic properties of 3d high-entropy alloys. Mater. Chem. Phys. 2018, 210, 320–326. [Google Scholar] [CrossRef]
- Zheng, S.-M.; Feng, W.-Q.; Wang, S.-Q. Elastic properties of high entropy alloys by MaxEnt approach. Comput. Mater. Sci. 2018, 142, 332–337. [Google Scholar] [CrossRef]
- Tian, F.; Wang, D.; Shen, J.; Wang, Y. An ab initio investgation of ideal tensile and shear strength of TiVNbMo high-entropy alloy. Mater. Lett. 2016, 166, 271–275. [Google Scholar] [CrossRef]
- Qiu, S.; Miao, N.; Zhou, J.; Guo, Z.; Sun, Z. Strengthening mechanism of aluminum on elastic properties of NbVTiZr high-entropy alloys. Intermetallics 2018, 92, 7–14. [Google Scholar] [CrossRef]
- Sharma, A.; Balasubramanian, G. Dislocation dynamics in Al0. 1CoCrFeNi high-entropy alloy under tensile loading. Intermetallics 2017, 91, 31–34. [Google Scholar] [CrossRef]
- Zaddach, A.; Niu, C.; Koch, C.; Irving, D. Mechanical properties and stacking fault energies of NiFeCrCoMn high-entropy alloy. JOM 2013, 65, 1780–1789. [Google Scholar] [CrossRef]
- Senkov, O.; Miller, J.; Miracle, D.; Woodward, C. Accelerated exploration of multi-principal element alloys with solid solution phases. Nat. Commun. 2015, 6, 6529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, M.; Belyea, D.; Bauer, C.; Bryant, N.; Michel, E.; Turgut, Z.; Leontsev, S.; Horwath, J.; Semiatin, S.; McHenry, M. Thermomagnetic analysis of FeCoCr x Ni alloys: Magnetic entropy of high-entropy alloys. J. Appl. Phys. 2013, 113, 17A923. [Google Scholar] [CrossRef]
- Dirras, G.; Lilensten, L.; Djemia, P.; Laurent-Brocq, M.; Tingaud, D.; Couzinié, J.-P.; Perrière, L.; Chauveau, T.; Guillot, I. Elastic and plastic properties of as-cast equimolar TiHfZrTaNb high-entropy alloy. Mater. Sci. Eng. A 2016, 654, 30–38. [Google Scholar] [CrossRef]
- Dirras, G.; Gubicza, J.; Heczel, A.; Lilensten, L.; Couzinié, J.-P.; Perrière, L.; Guillot, I.; Hocini, A. Microstructural investigation of plastically deformed Ti20Zr20Hf20Nb20Ta20 high entropy alloy by X-ray diffraction and transmission electron microscopy. Mater. Charact. 2015, 108, 1–7. [Google Scholar] [CrossRef]
- Štamborská, M.; Lapin, J. Effect of anisotropic microstructure on high-temperature compression deformation of CoCrFeNi based complex concentrated alloy. Kov. Mater. 2017, 55, 369–378. [Google Scholar]
- Lu, Y.; Dong, Y.; Jiang, L.; Wang, T.; Li, T.; Zhang, Y. A criterion for topological close-packed phase formation in high entropy alloys. Entropy 2015, 17, 2355–2366. [Google Scholar] [CrossRef]
- Ye, Y.; Zhang, Y.; He, Q.; Zhuang, Y.; Wang, S.; Shi, S.; Hu, A.; Fan, J.; Yang, Y. Atomic-scale distorted lattice in chemically disordered equimolar complex alloys. Acta Mater. 2018, 150, 182–194. [Google Scholar] [CrossRef]
- Ye, Y.; Liu, C.; Yang, Y. A geometric model for intrinsic residual strain and phase stability in high entropy alloys. Acta Mater. 2015, 94, 152–161. [Google Scholar] [CrossRef]
- Kivy, M.B.; Asle Zaeem, M. Generalized stacking fault energies, ductilities, and twinnabilities of CoCrFeNi-based face-centered cubic high entropy alloys. Scr. Mater. 2017, 139, 83–86. [Google Scholar] [CrossRef]
- Zhao, S.; Stocks, G.M.; Zhang, Y. Stacking fault energies of face-centered cubic concentrated solid solution alloys. Acta Mater. 2017, 134, 334–345. [Google Scholar] [CrossRef]
- Huang, S.; Li, W.; Lu, S.; Tian, F.; Shen, J.; Holmström, E.; Vitos, L. Temperature dependent stacking fault energy of FeCrCoNiMn high entropy alloy. Scr. Mater. 2015, 108, 44–47. [Google Scholar] [CrossRef]
- Pei, Z.; Eisenbach, M. Acceleration of the Particle Swarm Optimization for Peierls–Nabarro modeling of dislocations in conventional and high-entropy alloys. Comput. Phys. Commun. 2017, 215, 7–12. [Google Scholar] [CrossRef]
- Choudhuri, D.; Gwalani, B.; Gorsse, S.; Komarasamy, M.; Mantri, S.A.; Srinivasan, S.G.; Mishra, R.S.; Banerjee, R. Enhancing strength and strain hardenability via deformation twinning in fcc-based high entropy alloys reinforced with intermetallic compounds. Acta Mater. 2019, 165, 420–430. [Google Scholar] [CrossRef]
- Wang, Z.; Li, J.; Fang, Q.; Liu, B.; Zhang, L. Investigation into nanoscratching mechanical response of AlCrCuFeNi high-entropy alloys using atomic simulations. Appl. Surf. Sci. 2017, 416, 470–481. [Google Scholar] [CrossRef]
- Li, J.; Fang, Q.; Liu, B.; Liu, Y. Transformation induced softening and plasticity in high entropy alloys. Acta Mater. 2018, 147, 35–41. [Google Scholar] [CrossRef]
- Smith, T.; Hooshmand, M.; Esser, B.; Otto, F.; McComb, D.; George, E.; Ghazisaeidi, M.; Mills, M. Atomic-scale characterization and modeling of 60 dislocations in a high-entropy alloy. Acta Mater. 2016, 110, 352–363. [Google Scholar] [CrossRef]
- Zaddach, A.; Scattergood, R.; Koch, C. Tensile properties of low-stacking fault energy high-entropy alloys. Mater. Sci. Eng. A 2015, 636, 373–378. [Google Scholar] [CrossRef]
- Liu, S.; Wu, Y.; Wang, H.; He, J.; Liu, J.; Chen, C.; Liu, X.; Wang, H.; Lu, Z. Stacking fault energy of face-centered-cubic high entropy alloys. Intermetallics 2018, 93, 269–273. [Google Scholar] [CrossRef]
- Ventelon, L.; Lüthi, B.; Clouet, E.; Proville, L.; Legrand, B.; Rodney, D.; Willaime, F. Dislocation core reconstruction induced by carbon segregation in bcc iron. Phys. Rev. B 2015, 91, 220102. [Google Scholar] [CrossRef]
- Tsuru, T.; Chrzan, D. Effect of solute atoms on dislocation motion in Mg: An electronic structure perspective. Sci. Rep. 2015, 5, 8793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Q.; Qi, L.; Tsuru, T.; Traylor, R.; Rugg, D.; Morris, J.; Asta, M.; Chrzan, D.; Minor, A.M. Origin of dramatic oxygen solute strengthening effect in titanium. Science 2015, 347, 635–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varvenne, C.; Leyson, G.; Ghazisaeidi, M.; Curtin, W. Solute strengthening in random alloys. Acta Mater. 2017, 124, 660–683. [Google Scholar] [CrossRef] [Green Version]
- Varvenne, C.; Curtin, W.A. Strengthening of high entropy alloys by dilute solute additions: CoCrFeNiAlx and CoCrFeNiMnAlx alloys. Scr. Mater. 2017, 138, 92–95. [Google Scholar] [CrossRef]
- Toda-Caraballo, I.; Rivera-Díaz-del-Castillo, P.E. Modelling solid solution hardening in high entropy alloys. Acta Mater. 2015, 85, 14–23. [Google Scholar] [CrossRef]
- Toda-Caraballo, I. A general formulation for solid solution hardening effect in multicomponent alloys. Scr. Mater. 2017, 127, 113–117. [Google Scholar] [CrossRef]
- Walbrühl, M.; Linder, D.; Ågren, J.; Borgenstam, A. Modelling of solid solution strengthening in multicomponent alloys. Mater. Sci. Eng. A 2017, 700, 301–311. [Google Scholar] [CrossRef]
- Labusch, R. A statistical theory of solid solution hardening. Phys. Status Solidi 1970, 41, 659–669. [Google Scholar] [CrossRef]
- Toda-Caraballo, I.; Wróbel, J.; Dudarev, S.; Nguyen-Manh, D.; Rivera-Díaz-del-Castillo, P. Interatomic spacing distribution in multicomponent alloys. Acta Mater. 2015, 97, 156–169. [Google Scholar] [CrossRef]
- Huang, S.; Vida, Á.; Heczel, A.; Holmström, E.; Vitos, L. Thermal Expansion, Elastic and Magnetic Properties of FeCoNiCu-Based High-Entropy Alloys Using First-Principle Theory. JOM 2017, 69, 2107–2112. [Google Scholar] [CrossRef]
- Löffler, A.; Zendegani, A.; Gröbner, J.; Hampl, M.; Schmid-Fetzer, R.; Engelhardt, H.; Rettenmayr, M.; Körmann, F.; Hickel, T.; Neugebauer, J. Quaternary Al-Cu-Mg-Si Q Phase: Sample Preparation, Heat Capacity Measurement and First-Principles Calculations. J. Phase Equilib. Diffus. 2016, 37, 119–126. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, W.; Xu, Y.; Lu, Z.; Li, D. The thermal-mechanical behavior of WTaMoNb high-entropy alloy via selective laser melting (SLM): Experiment and simulation. Int. J. Adv. Manuf. Technol. 2018, 96, 461–474. [Google Scholar] [CrossRef]
- Rahul, M.; Samal, S.; Venugopal, S.; Phanikumar, G. Experimental and finite element simulation studies on hot deformation behaviour of AlCoCrFeNi2. 1 eutectic high entropy alloy. J. Alloy. Compd. 2018, 749, 1115–1127. [Google Scholar] [CrossRef]
- Zunger, A.; Wei, S.-H.; Ferreira, L.; Bernard, J.E. Special quasirandom structures. Phys. Rev. Lett. 1990, 65, 353. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Ding, X.; Feng, Y.; Liu, X.; Liu, K.; Lu, Z.; Li, D.; Li, Y.; Liu, C.; Chen, X.-Q. Vacancy formation enthalpies of high-entropy FeCoCrNi alloy via first-principles calculations and possible implications to its superior radiation tolerance. J. Mater. Sci. Technol. 2018, 34, 355–364. [Google Scholar] [CrossRef]
- Schneeweiss, O.; Friák, M.; Dudová, M.; Holec, D.; Šob, M.; Kriegner, D.; Holý, V.; Beran, P.; George, E.P.; Neugebauer, J. Magnetic properties of the CrMnFeCoNi high-entropy alloy. Phys. Rev. B 2017, 96, 014437. [Google Scholar] [CrossRef]
- Huang, S.; Holmström, E.; Eriksson, O.; Vitos, L. Mapping the magnetic transition temperatures for medium-and high-entropy alloys. Intermetallics 2018, 95, 80–84. [Google Scholar] [CrossRef]
- Körmann, F.; Ma, D.; Belyea, D.D.; Lucas, M.S.; Miller, C.W.; Grabowski, B.; Sluiter, M.H. “Treasure maps” for magnetic high-entropy-alloys from theory and experiment. Appl. Phys. Lett. 2015, 107, 142404. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Zhang, H.; Lu, S.; Ding, X.; Wang, Y.; Vitos, L. Phase selection rule for Al-doped CrMnFeCoNi high-entropy alloys from first-principles. Acta Mater. 2017, 140, 366–374. [Google Scholar] [CrossRef]
- Ma, S.; Zhang, S.; Gao, M.; Liaw, P.; Zhang, Y. A Successful Synthesis of the CoCrFeNiAl0. 3 Single-Crystal, High-Entropy Alloy by Bridgman Solidification. JOM 2013, 65, 1751–1758. [Google Scholar] [CrossRef]
- Zuo, T.; Gao, M.C.; Ouyang, L.; Yang, X.; Cheng, Y.; Feng, R.; Chen, S.; Liaw, P.K.; Hawk, J.A.; Zhang, Y. Tailoring magnetic behavior of CoFeMnNiX (X = Al, Cr, Ga, and Sn) high entropy alloys by metal doping. Acta Mater. 2017, 130, 10–18. [Google Scholar] [CrossRef]
- Calvo-Dahlborg, M.; Cornide, J.; Tobola, J.; Nguyen-Manh, D.; Wróbel, J.; Juraszek, J.; Jouen, S.; Dahlborg, U. Interplay of electronic, structural and magnetic properties as the driving feature of high-entropy CoCrFeNiPd alloys. J. Phys. D Appl. Phys. 2017, 50, 185002. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
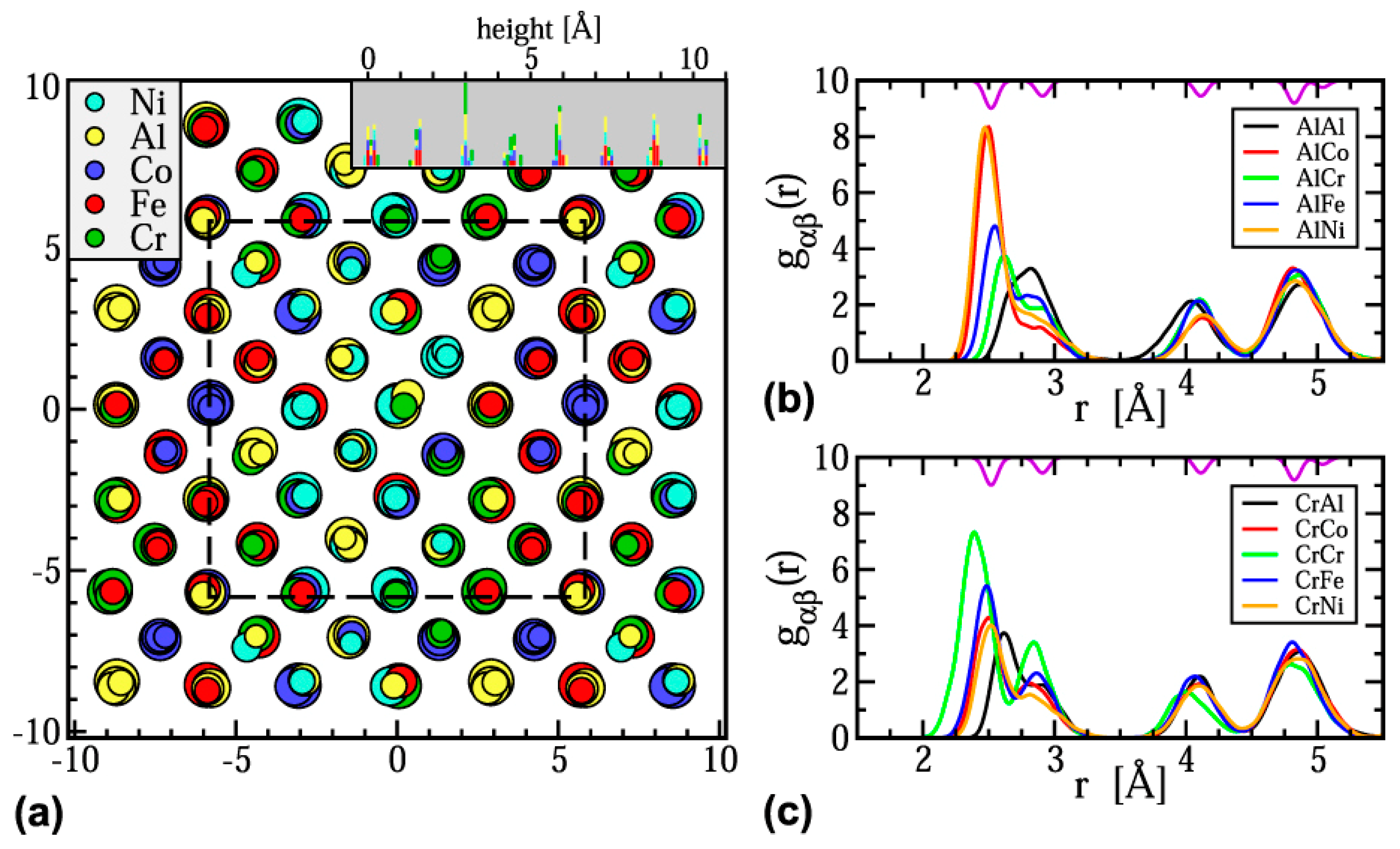{kind=link}
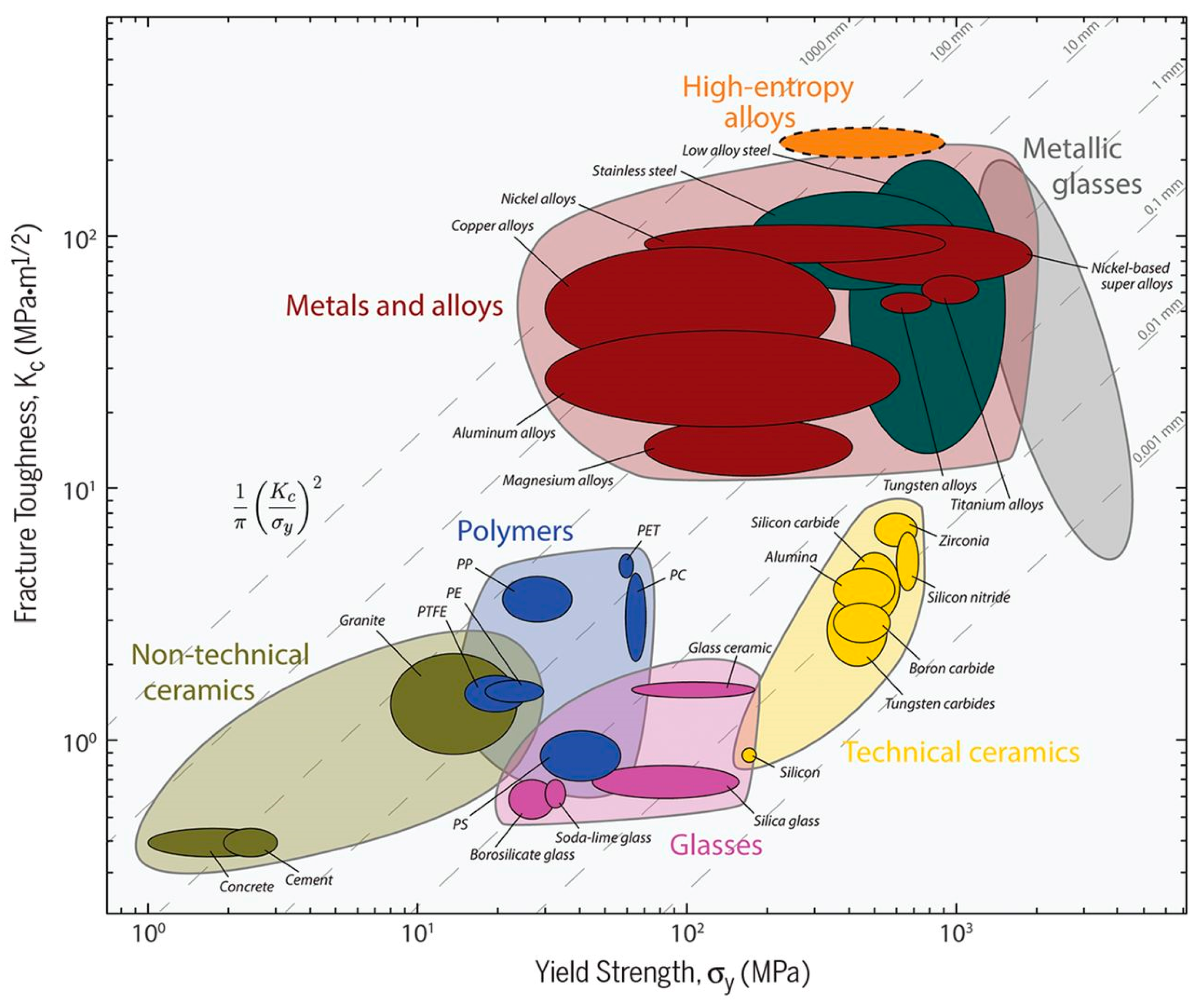{kind=link}
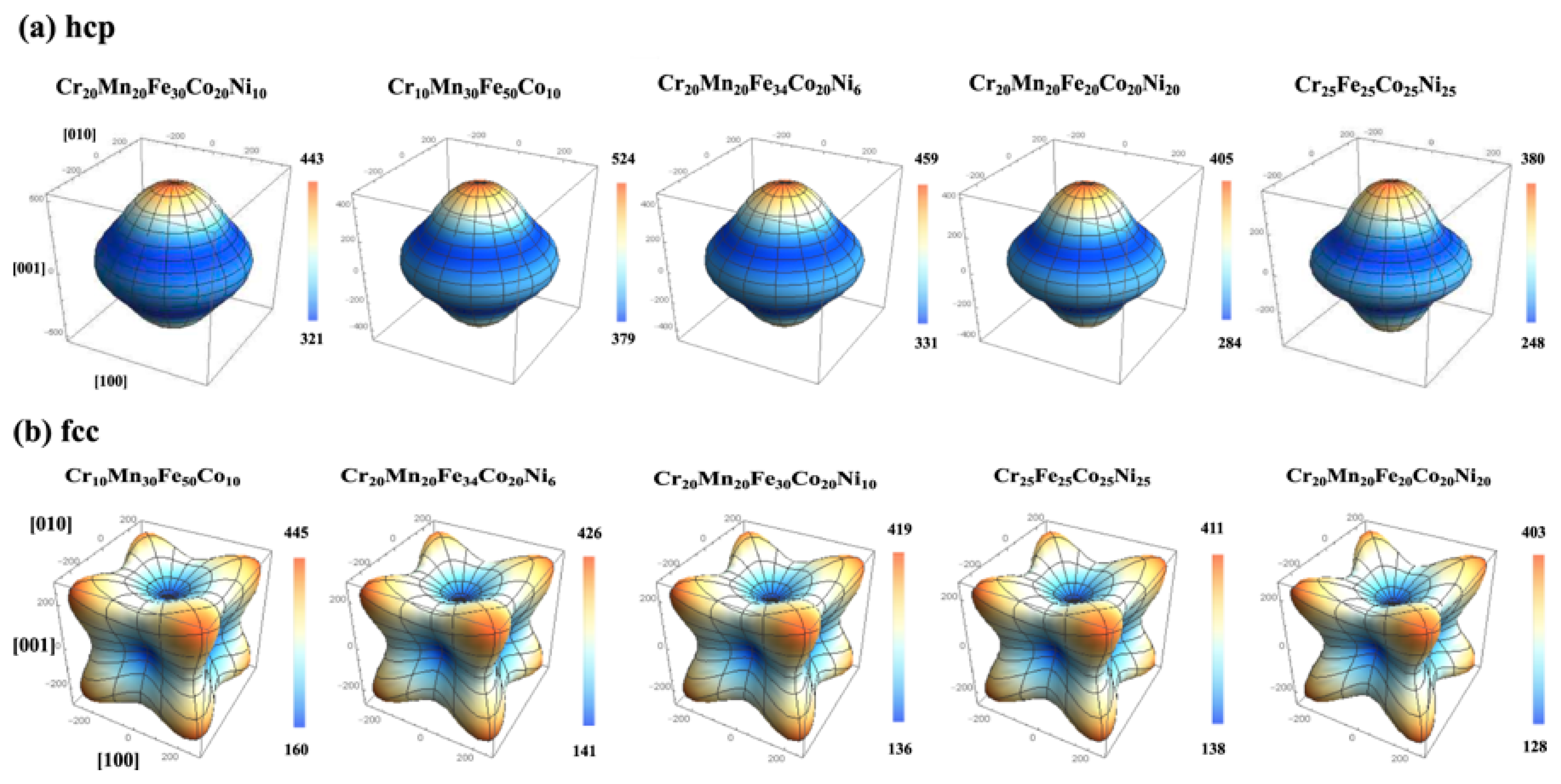{kind=link}
{kind=link}
{kind=link}
| Alloys | EDFT | EMD | Eexp. |
|---|---|---|---|
| Al0.1CoCrFeNi | - | 199 [99] | 203 [99] |
| CoCrFeNi | 275.7 [94], 274.1 [89], 225 [100], 196 [100] | - | 226 [101] |
| CoCrMnNi | 265.6 [89] | - | 171 [100] |
| CoFeMnNi | 267.2 [95] | - | - |
| CoCrFeMnNi | 262.4 [94], 279.7 [95], 207 [100] | - | 215 [102], 137 [100] |
| Cr10Mn40Fe40Co10 | 328.1 [94] | - | - |
| TiZrVMoTaNb | 71.9 [59] | - | - |
| TiZrVMoTaNbCr | 130.9 [59] | - | - |
| TiZrVMoTaNbCrW | 166.7 [59] | - | - |
| NbVTiZrAl | 118.0 [98] | - | - |
| ZrNbHf | 95.4 [93] | - | - |
| ZrVTiNb | 95.1 [98], 117.5 [93] | - | 80 [103], 101 [104] |
| ZrTiNbHf | 88.9 [93] | - | - |
| ZrVTiNbHf | 97.1 [93] | - | - |
| TiZrNbMoVx | 141.1 (x = 1) [90], 127.8 [96] | - | - |
| AlxMoNbTiV | 174.4 (x = 1) [92], 185.4 [96] | - | - |
| TaNbHfZrTi | 185.4 [96] | - | 78.5 [103], 87 [104] |
| NbTaTiWV | 257.3 [96] | - | - |
| WNbMoTaV | 218.0 [96] | - | - |
| MoNbTaTiV | 130.5 [96] | - | - |
| MoTiZrNbHfTa | 136.6 [96] | - | - |
| Alloys | γSFE-DFT | γSFE-MD | γSFE-exp |
|---|---|---|---|
| FeCrCoNiMn | 21 [111], 27.3 [100], 29.7 [109] | - | 25 [117], 26.5 [118], 19 [100] |
| CoCrFeNi | 31.6 [109], 31.7 [100] | - | 27 [118] |
| CoCrFeNiCu0.5 | 29.0 [109] | - | - |
| CoCrFeNiCu | 27.5 [109] | - | - |
| CoCrFeNiCuAl0.5 | 32.0 [109] | - | - |
| CoCrFeNiCuTi0.5 | 37.4 [109] | - | - |
| CoCrFeNiAl0.3 | 35.2 [109] | - | - |
| CoCrFeNi | 31.6 [109] | - | 26.8 [117] |
| CoCrFeNiCu0.5 | 29.0 [109] | - | - |
| CoCrFeNiCu | 27.5 [109] | - | 49.0 [115] |
| CoCrFeNiCuAl0.5 | 32.0 [109] | - | - |
| Co20Cr26Fe20Ni14Mn20 | - | - | 3.5 [117] |
| Co15Cr20Fe20Ni25Mn20 | - | - | 38 [118] |
| Co26Cr18.5Fe18.5Ni18.5Mn18.5 | - | - | 9.7 [100] |
| (CoCrFeNi)86Mn14 | - | - | 29 [118] |
| (CoCrFeNi)94Mn6 | - | - | 28 [118] |
| FeCrNi | - | 20 [114] | - |
| Method | First-Principles | Monte-Carlo | MD | Microscale (e.g., PFM simulations) | FEM | Thermodynamics | |
|---|---|---|---|---|---|---|---|
| Structures/ Phases | a | b | c | × | × | d | |
| Properties | Mechanical | e | × | f | × | g | × |
| Thermo-Chemical | h | × | × | × | i | × | |
| Magnetic | j | × | × | × | × | × | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beyramali Kivy, M.; Hong, Y.; Asle Zaeem, M. A Review of Multi-Scale Computational Modeling Tools for Predicting Structures and Properties of Multi-Principal Element Alloys. Metals 2019, 9, 254. https://doi.org/10.3390/met9020254
Beyramali Kivy M, Hong Y, Asle Zaeem M. A Review of Multi-Scale Computational Modeling Tools for Predicting Structures and Properties of Multi-Principal Element Alloys. Metals. 2019; 9(2):254. https://doi.org/10.3390/met9020254
Chicago/Turabian StyleBeyramali Kivy, Mohsen, Yu Hong, and Mohsen Asle Zaeem. 2019. "A Review of Multi-Scale Computational Modeling Tools for Predicting Structures and Properties of Multi-Principal Element Alloys" Metals 9, no. 2: 254. https://doi.org/10.3390/met9020254