
First-Principles Study of B Segregation at Austenite Grain Boundary and Its Effect on the Hardenability of Low-Alloy Steels
 and
and
Abstract
:1. Introduction
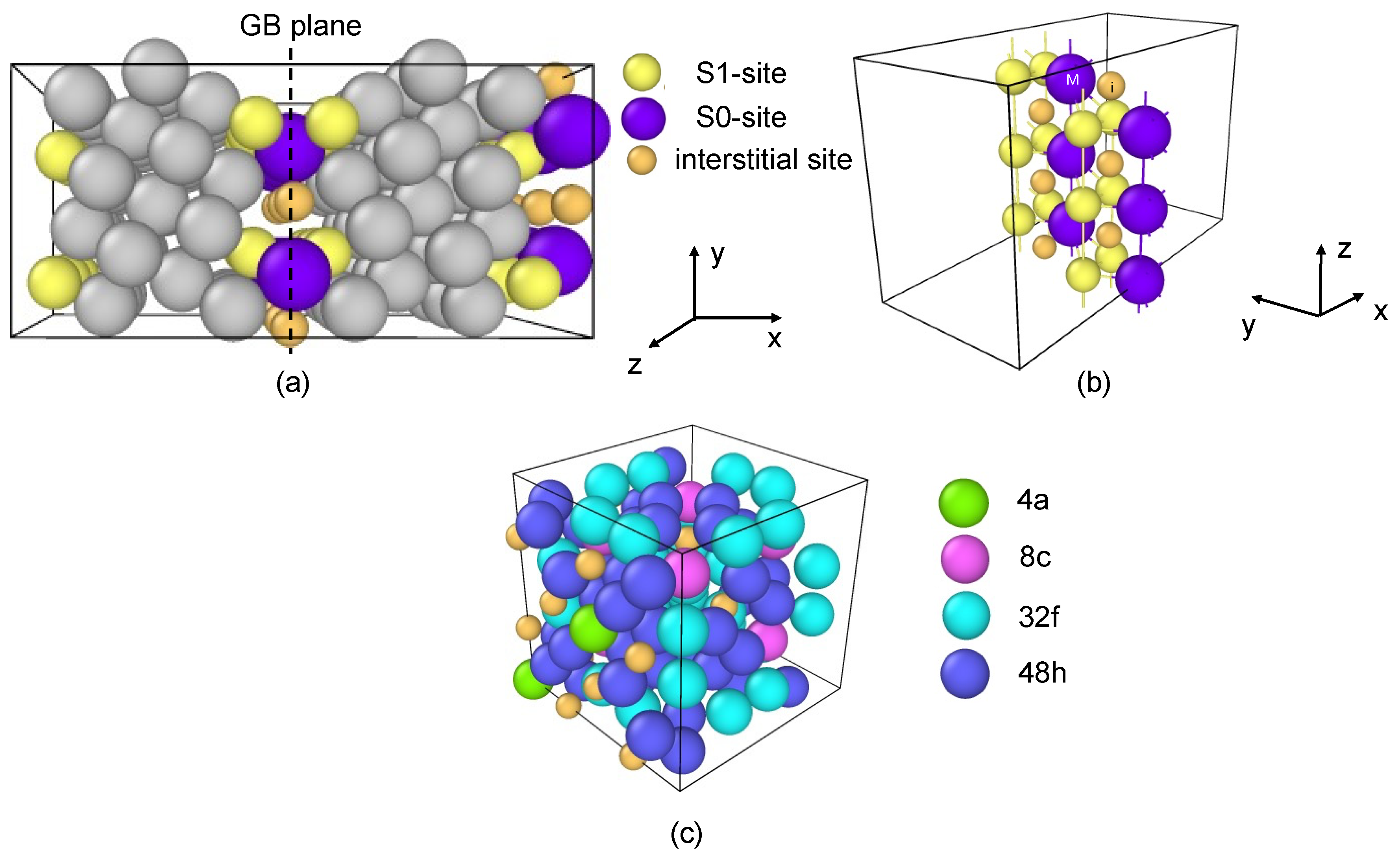
2. Methodology
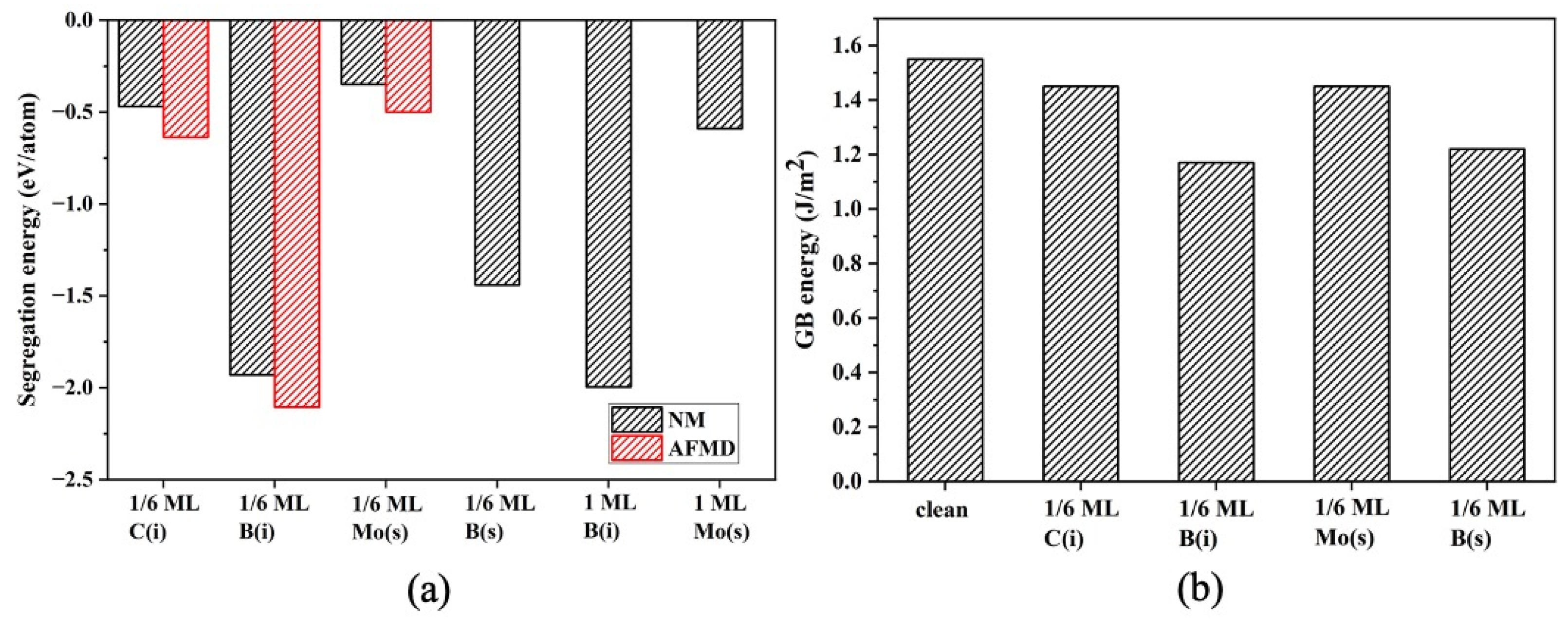
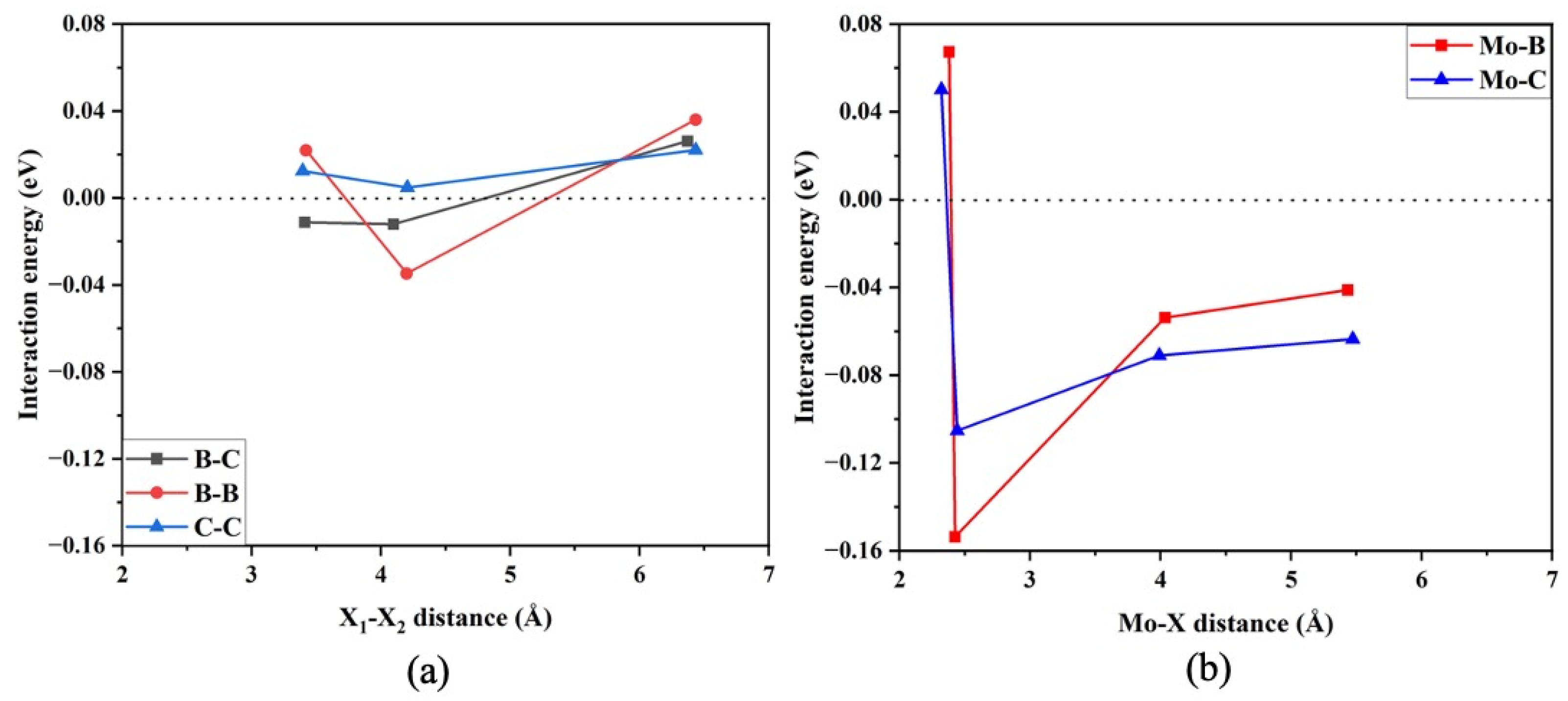
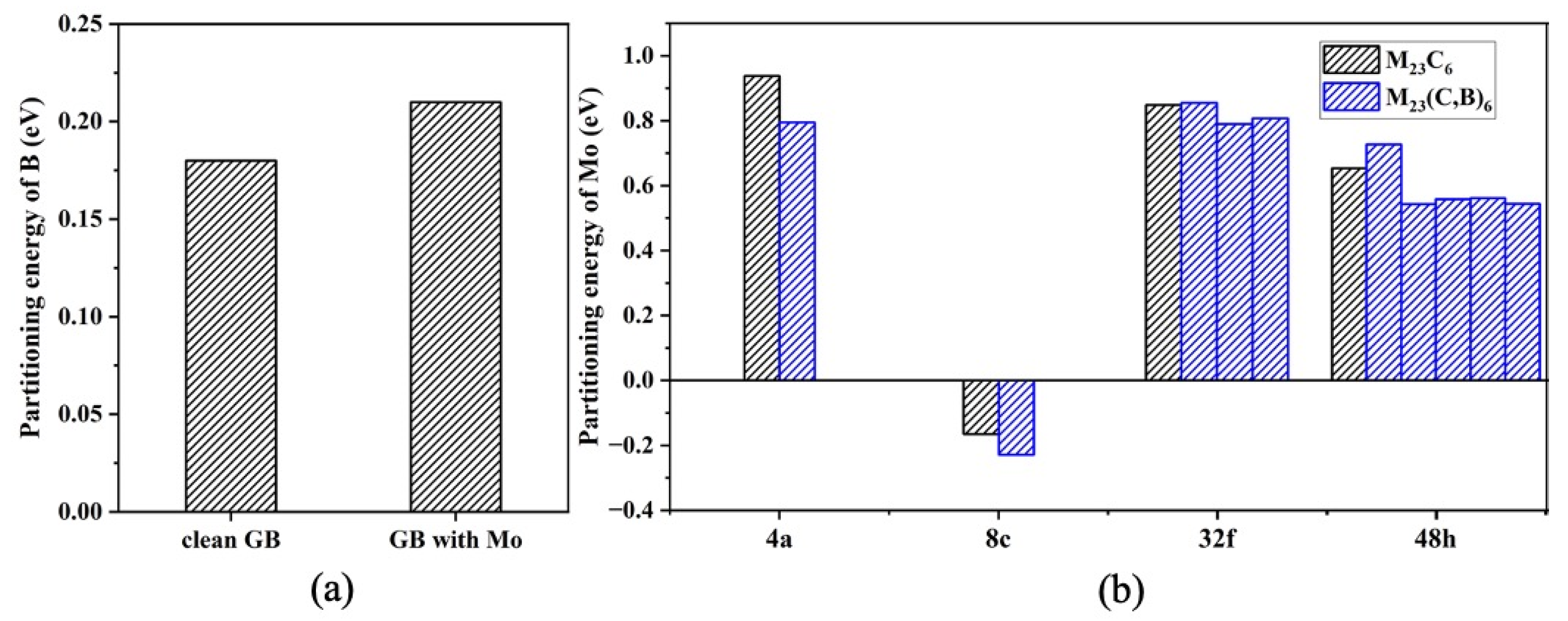
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aglan, H.; Liu, Z.; Hassan, M.; Fateh, M. Mechanical and fracture behavior of bainitic rail steel. J. Mater. Process. Technol. 2004, 151, 268–274. [Google Scholar] [CrossRef]
- Maitrepierre, P.; Rofes-Vernis, J.; Thivellier, D. Structure—Properties Relationships in Boron Steels, In Boron in Steels. In Proceedings of the International Symposium on Boron Steels, Milwaukee, WI, USA, 18 September 1979; The Metallurgical Society of AIME: New York, NY, USA, 1980; pp. 1–18. [Google Scholar]
- Taylor, K.; Hansen, S. The boron hardenability effect in thermomechanically processed, direct-quenched 0.2 Pct C steels. Metall. Trans. A 1990, 21, 1697–1708. [Google Scholar] [CrossRef]
- Ueno, M.; Inoue, T. Distribution of boron at austenite grain boundaries and bainitic transformation in low carbon steels. Trans. Iron Steel Inst. Jpn. 1973, 13, 210–217. [Google Scholar] [CrossRef]
- Morral, J.; Cameron, T. A model for ferrite nucleation applied to boron hardenability. Metall. Mater. A 1977, 8, 1817–1819. [Google Scholar] [CrossRef]
- Han, F.; Hwang, B.; Suh, D.-W.; Wang, Z.; Lee, D.L.; Kim, S.-J. Effect of molybdenum and chromium on hardenability of low-carbon boron-added steels. Met. Mater. Int. 2008, 14, 667–672. [Google Scholar] [CrossRef]
- Zhu, K.; Oberbillig, C.; Musik, C.; Loison, D.; Iung, T. Effect of B and B+ Nb on the bainitic transformation in low carbon steels. Mater. Sci. Eng. A 2011, 528, 4222–4231. [Google Scholar] [CrossRef]
- Asahi, H. Effects of Mo addition and austenitizing temperature on hardenability of low alloy B-added steels. ISIJ Int. 2002, 42, 1150–1155. [Google Scholar] [CrossRef]
- Hara, T.; Asahi, H.; Uemori, R.; Tamehiro, H. Role of combined addition of niobium and boron and of molybdenum and boron on hardnenability in low carbon steels. ISIJ Int. 2004, 44, 1431–1440. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, C.; Zhang, Z.; Dong, N.; Wang, J.; Liu, Y.; Lei, Z.; Han, P. Effects of B on the Segregation Behavior of Mo at the Fe–Cr (111)/Cr2O3 (0001) Interface: A First-Principles Study. Metals 2020, 10, 577. [Google Scholar] [CrossRef]
- Li, J.; Zhang, C.; Xu, L.; Zhang, Z.; Dong, N.; Liu, Y.; Wang, J.; Zhang, Y.; Ling, L.; Han, P. Effects of B on the segregation of Mo at the Fe-Cr-NiΣ5 (210) grain boundary. Phys. B Condens. Matter 2019, 568, 25–30. [Google Scholar] [CrossRef]
- Wang, J.; Enomoto, M.; Shang, C. First-principles study on the P-induced embrittlement and de-embrittling effect of B and C in ferritic steels. Acta Mater. 2021, 219, 117260. [Google Scholar] [CrossRef]
- He, S.; Scheiber, D.; Jechtl, T.; Moitzi, F.; Peil, O.; Romaner, L.; Zamberger, S.; Povoden-Karadeniz, E.; Razumovskiy, V.; Ruban, A.V. Solubility and segregation of B in paramagnetic fcc Fe. Phys. Rev. Mater. 2022, 6, 23604. [Google Scholar] [CrossRef]
- Li, Y.; Han, C.; Zhang, C.; Jia, K.; Han, P.; Wu, X. Effects of alloying on the behavior of B and S at Σ5 (2 1 0) grain boundary in γ-Fe. Comput. Mater. Sci. 2016, 115, 170–176. [Google Scholar] [CrossRef] [Green Version]
- Sawada, H. First-principles study of grain boundary embrittlement in Fe–Ni–S alloy. Comput. Mater. Sci. 2012, 55, 17–22. [Google Scholar] [CrossRef]
- Wang, J.; Enomoto, M.; Guo, H.; Shang, C. First-principles study on the equilibrium shape of nanometer-sized body-centered cubic Cu precipitates in ferritic steels. Comput. Mater. Sci. 2020, 172, 109351. [Google Scholar] [CrossRef]
- Wang, J.; Enomoto, M.; Shang, C. First-principles study on the interfacial segregation at coherent Cu precipitate/Fe matrix interface. Scripta Mater. 2020, 185, 42–46. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.E.; Carter, E.A. Carbon dissolution and diffusion in ferrite and austenite from first principles. Phys. Rev. B 2003, 67, 214103. [Google Scholar] [CrossRef] [Green Version]
- Jin, H. Atomistic Simulations of Solute-Interface Interactions in Iron. Ph.D. Thesis, University of British Columbia, Vancouver, BC, Canada, 2014. [Google Scholar]
- Ito, K.; Tanaka, Y.; Mitsunobu, T.; Kohtake, T.; Tsutsui, K.; Sawada, H. First-principles computational tensile test of γ-Fe grain boundaries considering the effect of magnetism: Electronic origin of grain boundary embrittlement due to Zn segregation. Phys. Rev. Mater. 2022, 6, 053604. [Google Scholar] [CrossRef]
- Hickel, T.; Nazarov, R.; McEniry, E.; Leyson, G.; Grabowski, B.; Neugebauer, J. Ab initio based understanding of the segregation and diffusion mechanisms of hydrogen in steels. JOM 2014, 66, 1399–1405. [Google Scholar] [CrossRef]
- Ye, F.; Tong, K.; Wang, Y.K.; Li, Z.; Zhou, F. First-principles study of interaction between vacancies and nitrogen atoms in fcc iron. Comput. Mater. Sci. 2018, 149, 65–72. [Google Scholar] [CrossRef]
- Ismer, L.; Hickel, T.; Neugebauer, J. Ab initio study of the solubility and kinetics of hydrogen in austenitic high Mn steels. Phys. Rev. B 2010, 81, 094111. [Google Scholar] [CrossRef]
- Fang, C.; Van Huis, M.; Sluiter, M. Formation, structure and magnetism of the γ-(Fe, M) 23C6 (M = Cr, Ni) phases: A first-principles study. Acta Mater. 2016, 103, 273–279. [Google Scholar] [CrossRef]
- Ande, C.K.; Sluiter, M.H. First-principles prediction of partitioning of alloying elements between cementite and ferrite. Acta Mater. 2010, 58, 6276–6281. [Google Scholar] [CrossRef]
- Ponomareva, A.; Gornostyrev, Y.N.; Abrikosov, I. Ab initio calculation of the solution enthalpies of substitutional and interstitial impurities in paramagnetic fcc Fe. Phys. Rev. B 2014, 90, 014439. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Ponge, D.; Choi, P.-P.; Raabe, D. Segregation of boron at prior austenite grain boundaries in a quenched martensitic steel studied by atom probe tomography. Scripta Mater. 2015, 96, 13–16. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Supercell | Number of Atoms | k-Point Mesh |
|---|---|---|
| 1 × 1 × 3 GB | 120 | 4 × 2 × 4 |
| 1 × 1 × 2 GB | 80 | 4 × 2 × 6 |
| M23(C,B)6 | 116 | 4 × 4 × 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Yang, X.; Qian, R.; Rong, X.; Xie, Z.; Shang, C. First-Principles Study of B Segregation at Austenite Grain Boundary and Its Effect on the Hardenability of Low-Alloy Steels. Metals 2022, 12, 2006. https://doi.org/10.3390/met12122006
Wang J, Yang X, Qian R, Rong X, Xie Z, Shang C. First-Principles Study of B Segregation at Austenite Grain Boundary and Its Effect on the Hardenability of Low-Alloy Steels. Metals. 2022; 12(12):2006. https://doi.org/10.3390/met12122006
Chicago/Turabian StyleWang, Jingliang, Xiang Yang, Rongtao Qian, Xuequan Rong, Zhenjia Xie, and Chengjia Shang. 2022. "First-Principles Study of B Segregation at Austenite Grain Boundary and Its Effect on the Hardenability of Low-Alloy Steels" Metals 12, no. 12: 2006. https://doi.org/10.3390/met12122006