
A Mechanistic Study of Clustering and Diffusion of Molybdenum and Rhenium Atoms in Liquid Sodium


Abstract
:1. Introduction
2. Computational Methods
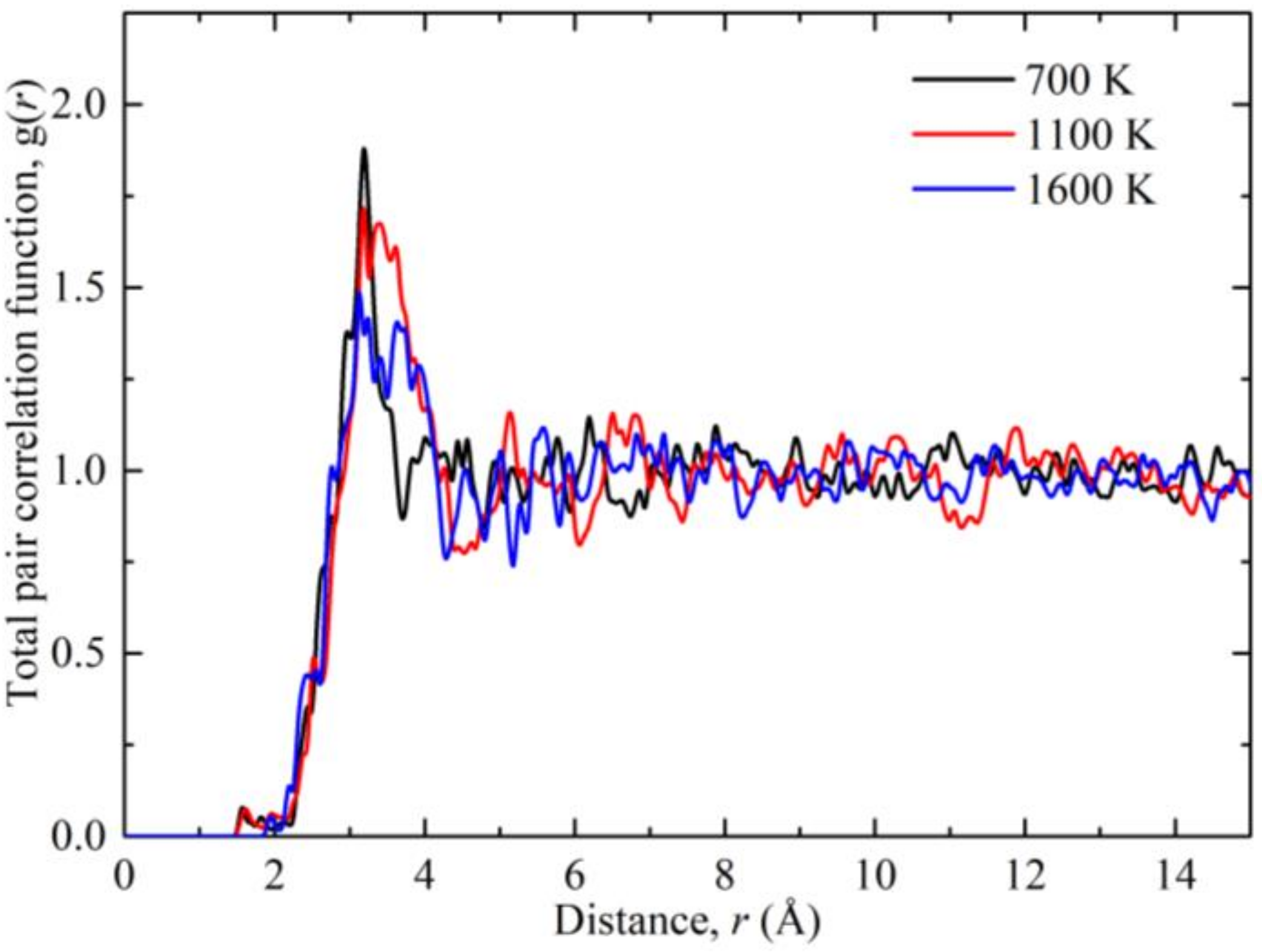
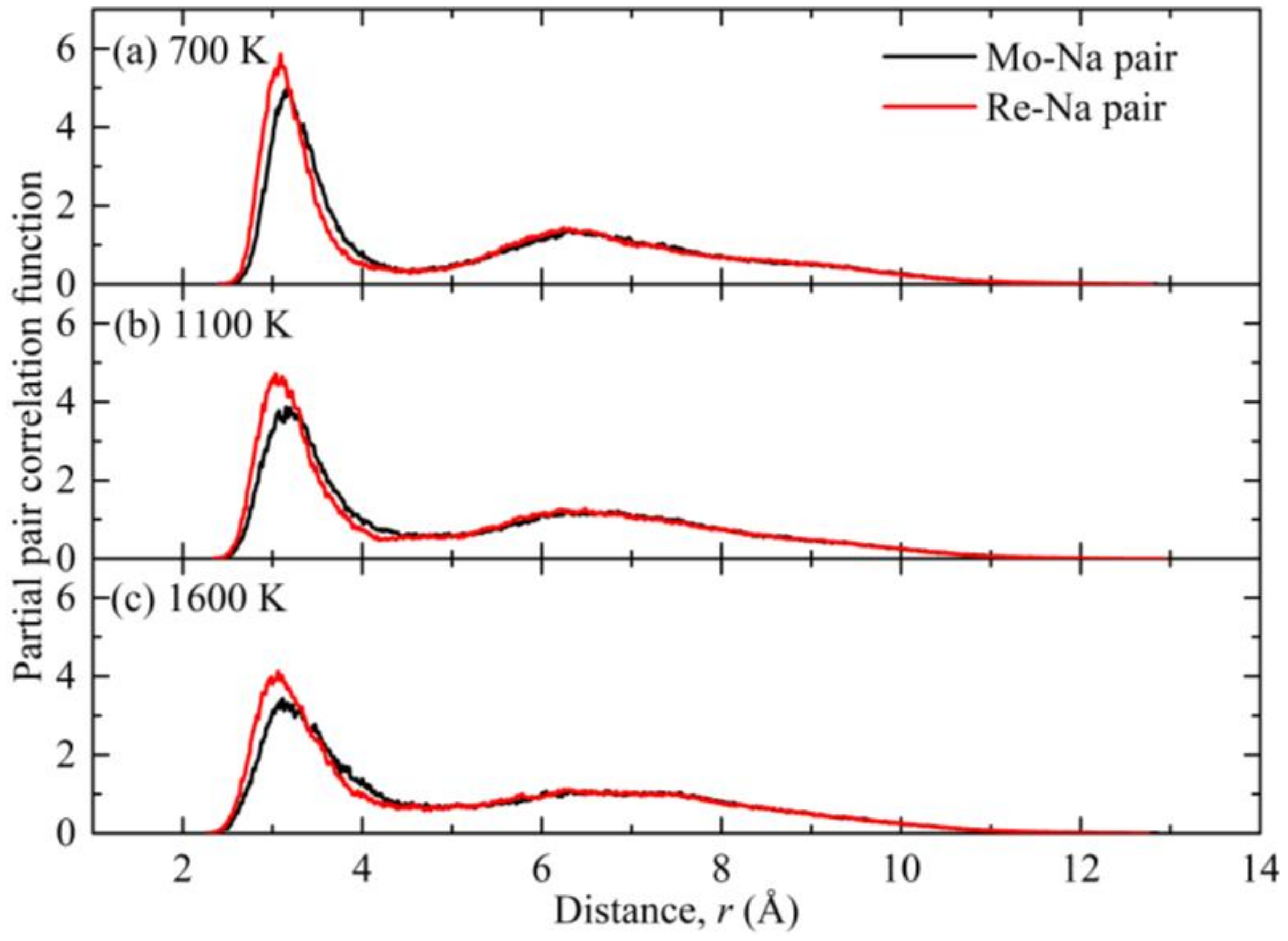
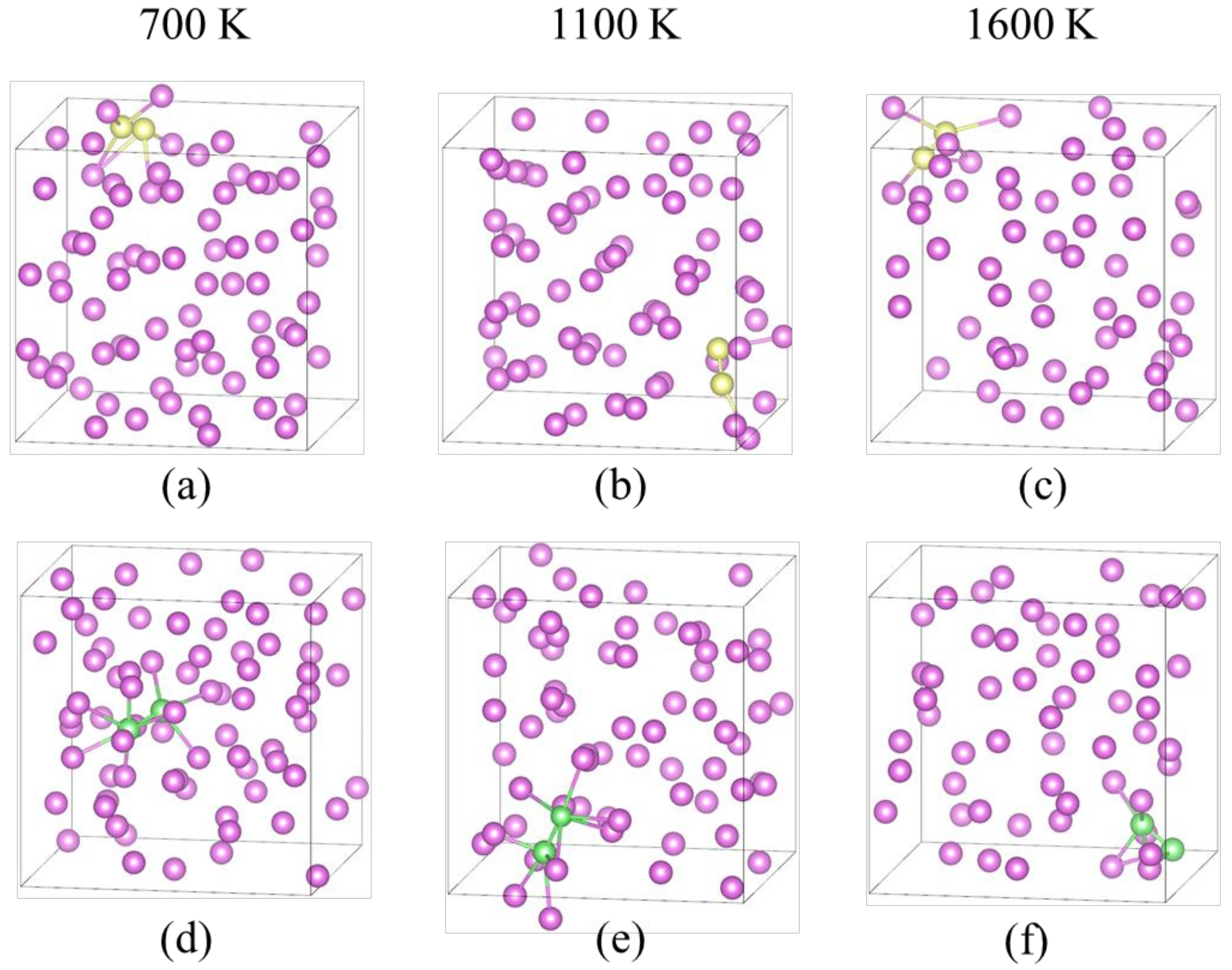
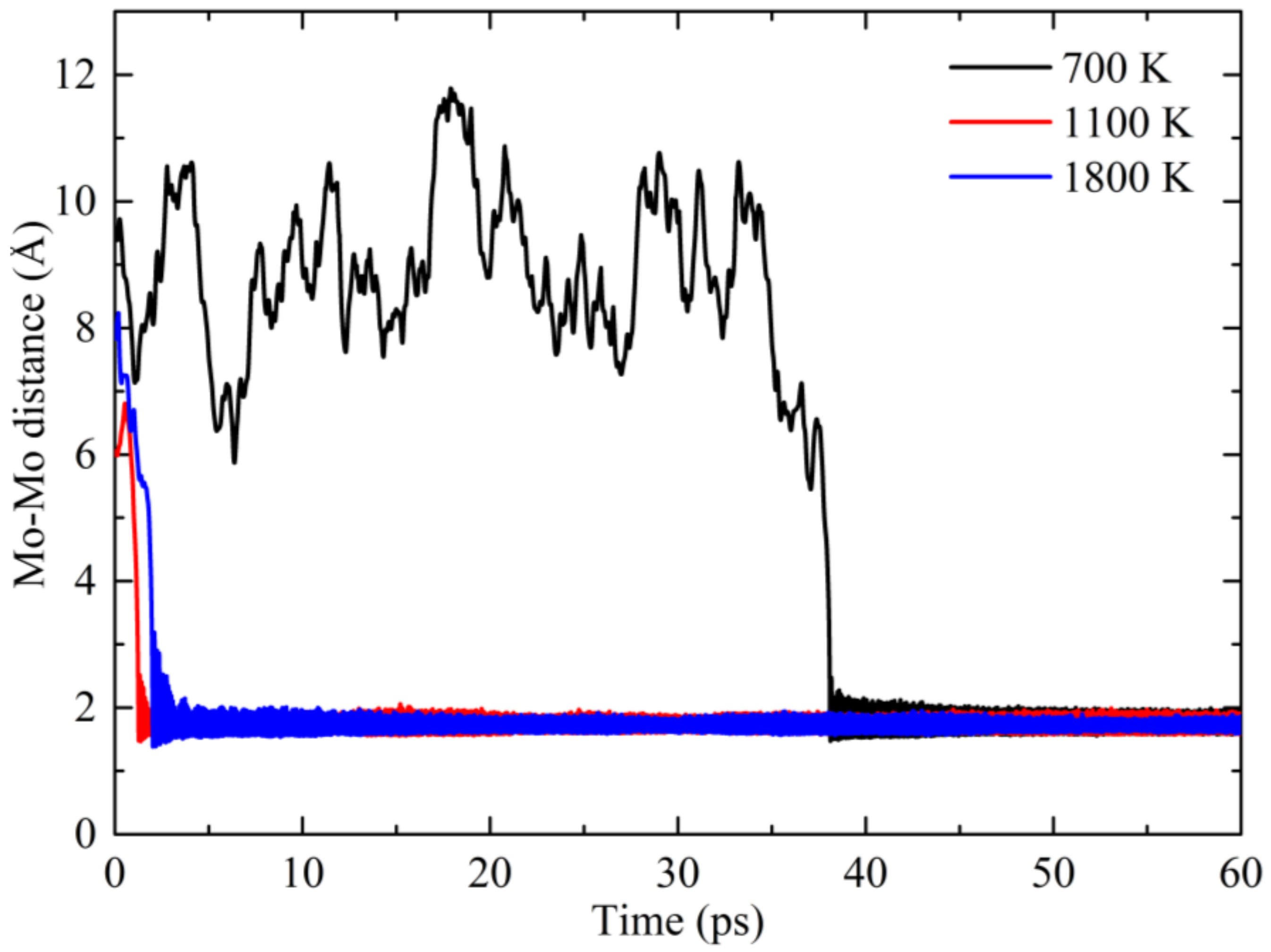
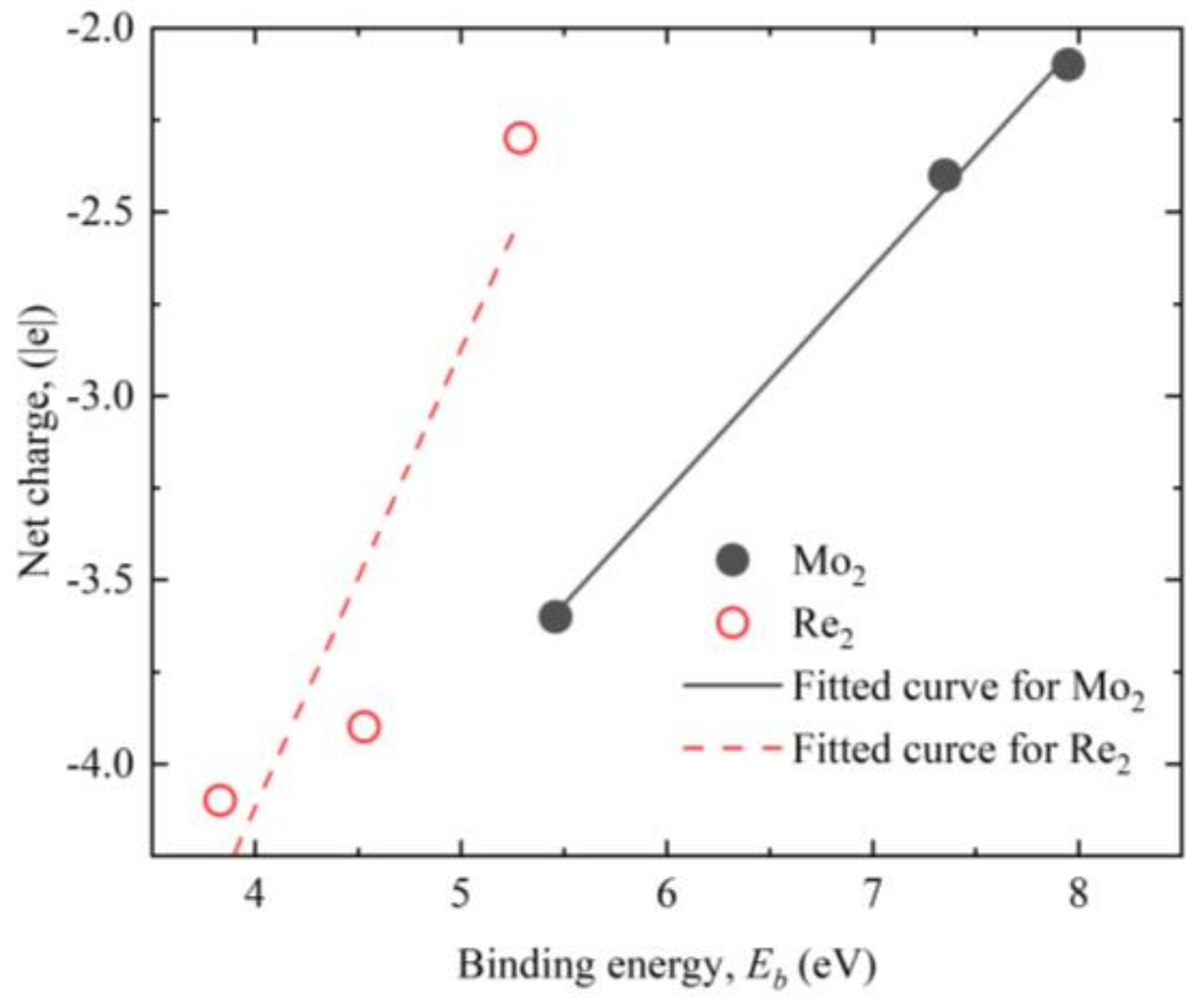
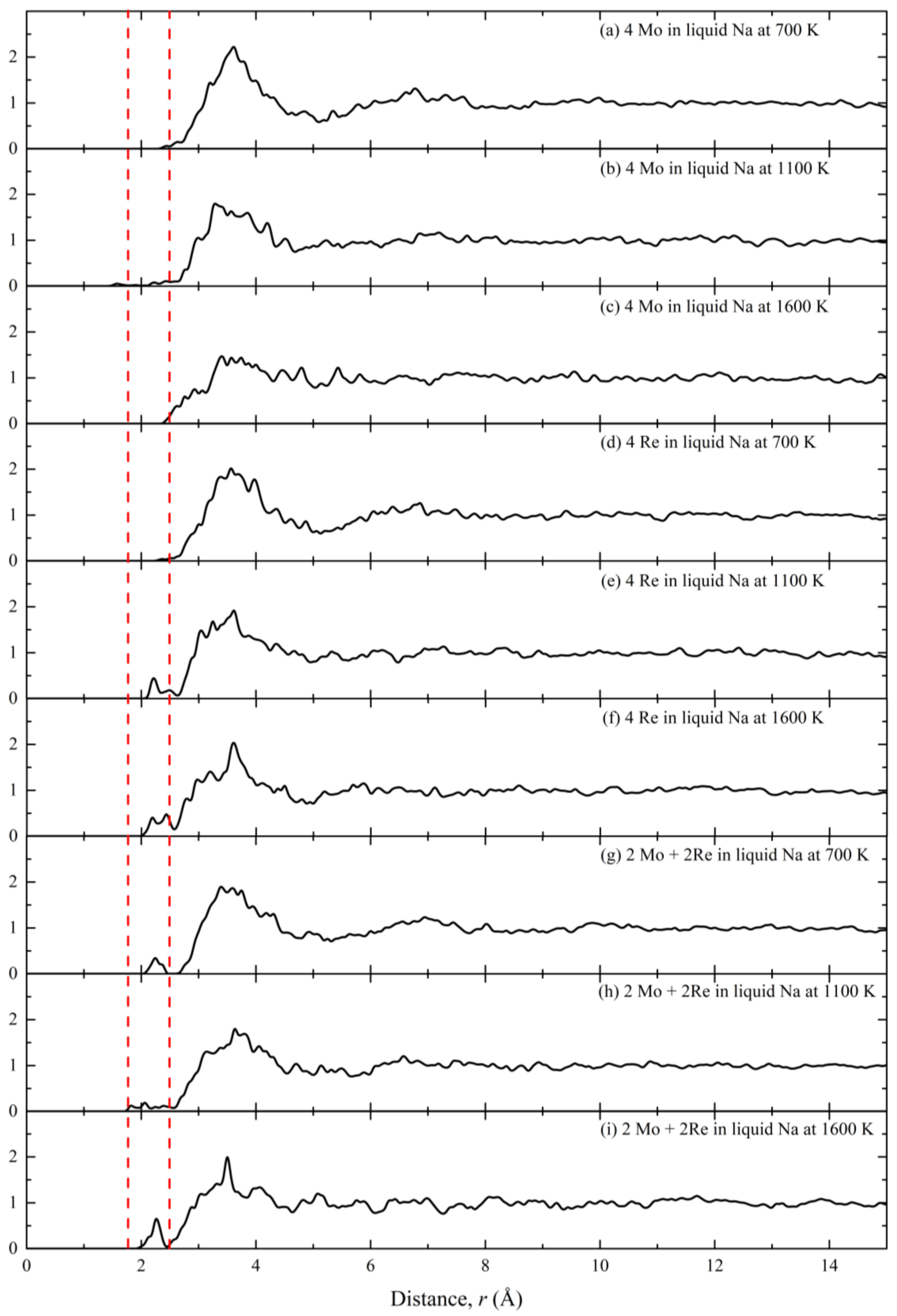
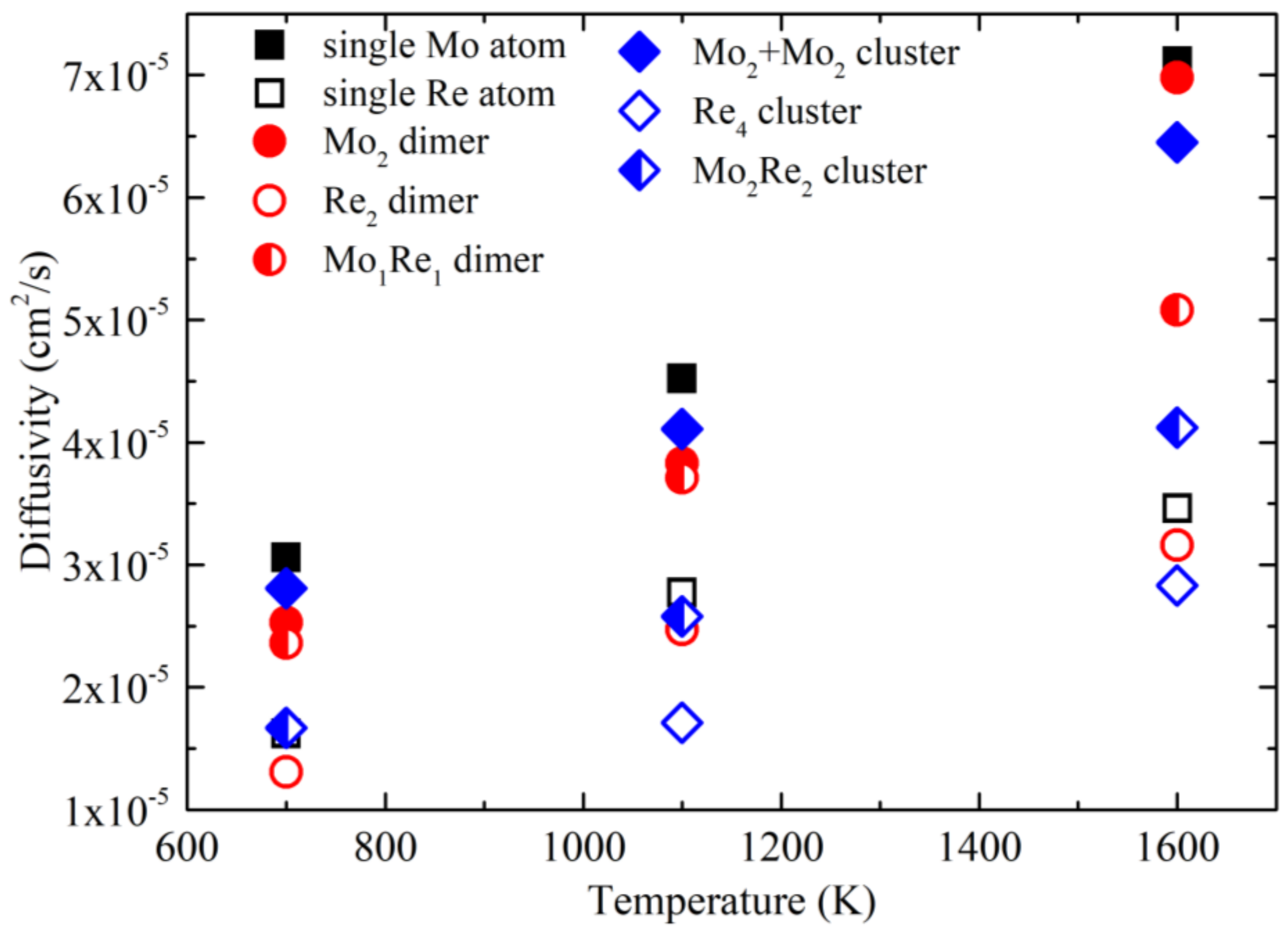
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tachan, Z.; Rühle, S.; Zaban, A. Dye-sensitized solar tubes: A new solar cell design for efficient current collection and im-proved cell sealing. Sol. Energy Mater. Sol. Cells 2010, 94, 317–322. [Google Scholar] [CrossRef]
- Zhou, L. Progress and problems in hydrogen storage methods. Renew. Sustain. Energy Rev. 2005, 9, 395–408. [Google Scholar] [CrossRef]
- Kim, T.; Song, W.; Son, D.-Y.; Ono, L.K.; Qi, Y. Lithium-ion batteries: Outlook on present, future, and hybridized technologies. J. Mater. Chem. A 2019, 7, 2942–2964. [Google Scholar] [CrossRef]
- Foley, A.M.; Leahy, P.G.; Marvuglia, A.; Mckeogh, E.J. Current methods and advances in forecasting of wind power genera-tion. Renew. Energy 2012, 37, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Ion, S. Nuclear energy: Current situation and prospects to 2020, Philosophical Transactions of the Royal Society a-Mathematical. Phys. Eng. Sci. 2007, 365, 935–944. [Google Scholar]
- Nifenecker, H. Future electricity production methods. Part 1: Nuclear energy. Rep. Prog. Phys. 2011, 74, 022801. [Google Scholar] [CrossRef]
- Pravalie, R.; Bandoc, G. Nuclear energy: Between global electricity demand, worldwide decarbonisation imperativeness, and planetary environmental implications. J. Environ. Manag. 2018, 209, 81–92. [Google Scholar] [CrossRef]
- El-Genk, M.S. Deployment history and design considerations for space reactor power systems. Acta Astronaut. 2009, 64, 833–849. [Google Scholar] [CrossRef]
- Li, Z.; Yang, X.; Wang, J.; Zhang, Z. Off-design performance and control characteristics of space reactor closed Brayton cycle system. Ann. Nucl. Energy 2019, 128, 318–329. [Google Scholar] [CrossRef]
- Grover, G.M.; Cotter, T.P.; Erickson, G.F. Structures of Very High Thermal Conductance. J. Appl. Phys. 1964, 35, 1990–1991. [Google Scholar] [CrossRef]
- Leonhardt, T.; Carlén, J.; Buck, M.; Brinkman, C.R.; Stevens, C.O. Investigation of mechanical properties and microstructure of various molybdenum-rhenium alloys. AIP Conf. Proc. 1999, 458, 685–690. [Google Scholar] [CrossRef]
- Busby, J.; Leonard, K.; Zinkle, S. Radiation-damage in molybdenum–rhenium alloys for space reactor applications. J. Nucl. Mater. 2007, 366, 388–406. [Google Scholar] [CrossRef]
- El-Genk, M.S.; Tournier, J.-M. A review of refractory metal alloys and mechanically alloyed-oxide dispersion strengthened steels for space nuclear power systems. J. Nucl. Mater. 2005, 340, 93–112. [Google Scholar] [CrossRef]
- Inoue, S.; Kano, S.; Saito, J.-I.; Isshiki, Y.; Yoshida, E.; Morinaga, M. Corrosion Behaviour of Nb-Based and Mo-Based Alloys in Liquid Na. In Liquid Metal Systems; Springer: Berlin/Heidelberg, Germany, 1995; pp. 75–83. [Google Scholar] [CrossRef]
- Saito, J.-I.; Inoue, S.; Kano, S.; Yuzawa, T.; Furui, M.; Morinaga, M. Alloying effects on the corrosion behavior of binary Nb-based and Mo-based alloys in liquid Li. J. Nucl. Mater. 1999, 264, 216–227. [Google Scholar] [CrossRef]
- Tu, S.-T.; Zhang, H.; Zhou, W.-W. Corrosion failures of high temperature heat pipes. Eng. Fail. Anal. 1999, 6, 363–370. [Google Scholar] [CrossRef]
- Yoshida, E.; Furukawa, T. Corrosion issues in sodium-cooled fast reactor (SFR) systems. In Nuclear Corrosion Science and Engineering; Woodhead Publishing: Sawston, UK, 2012; pp. 773–806. [Google Scholar] [CrossRef]
- Brissonneau, L. New considerations on the kinetics of mass transfer in sodium fast reactors: An attempt to consider irradia-tion effects and low temperature corrosion. J. Nucl. Mater. 2012, 423, 67–78. [Google Scholar] [CrossRef]
- Koci, L.; Ahuja, R.; Vitos, L.; Pinsook, U. Melting of Na at high pressure from ab initio calculations. Phys. Rev. B 2008, 77, 132101. [Google Scholar] [CrossRef]
- Yuryev, A.A.; Gelchinski, B.R. Simulation of properties of liquid alkali metals at high temperatures and pressures by ab initio molecular dynamics method. Dokl. Phys. 2015, 60, 105–108. [Google Scholar] [CrossRef]
- Li, X.; Samin, A.; Zhang, J.; Unal, C.; Mariani, R. Ab-initio molecular dynamics study of lanthanides in liquid sodium. J. Nucl. Mater. 2016, 484, 98–102. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio. Phys. Rev. B 1993, 48, 13115–13118. [Google Scholar] [CrossRef]
- Car, R.; Parrinello, M. Unified Approach for Molecular Dynamics and Density-Functional Theory. Phys. Rev. Lett. 1985, 55, 2471–2474. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.O.; Gunnarsson, O. The density functional formalism, its applications and prospects. Rev. Mod. Phys. 1989, 61, 689–746. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, J.; Leibowitz, L. Thermodynamic and Transport Properties of Sodium Liquid and Vapor; Argonne National Laboratory: Argonne, IL, USA, 1995. [CrossRef] [Green Version]
- Wang, V.; Xu, N.; Liu, J.-C.; Tang, G.; Geng, W.-T. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 2021, 267, 108033. [Google Scholar] [CrossRef]
- Winter, R.; Hensel, F.; Bodensteiner, T.; Gläser, W. The static structure factor of cesium over the whole liquid range up to the critical point. Z. Elektrochem. Ber. Bunsenges. Phys. Chem. 2015, 91, 1327–1330. [Google Scholar] [CrossRef]
- Bickham, S.R.; Pfaffenzeller, O.; Collins, L.A.; Kress, J.D.; Hohl, D. Ab initio molecular dynamics of expanded liquid sodium. Phys. Rev. B 1998, 58, R11813–R11816. [Google Scholar] [CrossRef]
- Meyer, R.E.; Nachtrieb, N.H. Self-Diffusion of Liquid Sodium. J. Chem. Phys. 1955, 23, 1851–1854. [Google Scholar] [CrossRef]
- Tang, W.; Sanville, E.; Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter Inst. Phys. J. 2009, 21, 084204. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature (K) | ||
|---|---|---|
| Mo2 | Re2 | |
| 700 | 5.46 | 3.83 |
| 1100 | 7.35 | 4.53 |
| 1600 | 7.95 | 5.29 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Ma, M.; Liang, W.; Deng, H. A Mechanistic Study of Clustering and Diffusion of Molybdenum and Rhenium Atoms in Liquid Sodium. Metals 2021, 11, 1430. https://doi.org/10.3390/met11091430
Liu Z, Ma M, Liang W, Deng H. A Mechanistic Study of Clustering and Diffusion of Molybdenum and Rhenium Atoms in Liquid Sodium. Metals. 2021; 11(9):1430. https://doi.org/10.3390/met11091430
Chicago/Turabian StyleLiu, Zhixiao, Mingyang Ma, Wenfeng Liang, and Huiqiu Deng. 2021. "A Mechanistic Study of Clustering and Diffusion of Molybdenum and Rhenium Atoms in Liquid Sodium" Metals 11, no. 9: 1430. https://doi.org/10.3390/met11091430