Effect of Sign-Alternating Cyclic Polarisation and Hydrogen Uptake on the Localised Corrosion of X70 Pipeline Steel in Near-Neutral Solutions

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- CEN-EN 15280. Evaluation of A.C. corrosion likelihood of buried pipelines-Application to cathodically protected pipelines. In Technical Specification; BSI: London, UK, 2013. [Google Scholar]
- Peabody, A.W. Peabody’s Control of Pipeline Corrosion, 2nd ed.; NACE International the Corrosion Society: Houston, TX, USA, 2001; pp. 226–231. [Google Scholar]
- Baeckmann, W.; Schwenk, W. Handbuch des kathodischen Korrosionsschutzes. In Theorie und Praxis der Elektrochemischen Schutzverfahren; Verlag Chemie: Weinheim, Germany, 1980; pp. 15–61. [Google Scholar]
- Lalvani, S.B.; Lin, X.A. A theoretical approach for predicting AC-induced corrosion. Corros. Sci. 1994, 6, 1039–1046. [Google Scholar] [CrossRef]
- Lalvani, S.B.; Lin, X.A. A revised model for predicting corrosion of materials induced by alternating voltages. Corros. Sci. 1996, 10, 1709–1719. [Google Scholar] [CrossRef]
- Bosch, R.W.; Bogaerts, W.F. A theoretical study of AC-induced corrosion considering diffusion phenomena. Corros. Sci. 1998, 2, 323–336. [Google Scholar] [CrossRef]
- Lalvani, S.B.; Zhang, G. The corrosion of carbon steel in a chloride environment due to periodic voltage modulation: Part I. Corros. Sci. 1995, 10, 1567–1582. [Google Scholar] [CrossRef]
- Fu, A.Q.; Cheng, Y.F. Effects of alternating current on corrosion of a coated pipeline steel in a chloride-containing carbonate/bicarbonate solution. Corros. Sci. 2010, 2, 612–619. [Google Scholar] [CrossRef]
- Goidanich, S.; Lazzari, L.; Ormellese, M. AC corrosion. Part 2: Parameters influencing corrosion rate. Corros. Sci. 2010, 3, 916–922. [Google Scholar] [CrossRef]
- Xu, L.Y.; Su, X.; Yin, Z.X.; Tang, Y.H.; Cheng, Y.F. Development of a real-time AC/DC data acquisition technique for studies of AC corrosion of pipelines. Corros. Sci. 2012, 61, 215–223. [Google Scholar] [CrossRef]
- Marshakov, A.I.; Nenasheva, T.A. The effect of alternating current on rate of dissolution of carbon steel in chloride electrolyte. Part I. Conditions of free corrosion. Prot. Met. Phys. Chem. Surf. 2017, 7, 1214–1221. [Google Scholar] [CrossRef]
- Kim, D.-K.; Muralidharan, S.; Ha, T.-H.; Bae, J.-H.; Ha, Y.-C.; Lee, H.-G.; Scantlebury, J.D. Electrochemical studies on the alternating current corrosion of mild steel under cathodic protection condition in marine environments. Electrochim. Acta 2006, 25, 5259–5267. [Google Scholar] [CrossRef]
- Kuang, D.; Cheng, Y.F. Understand the AC induced pitting corrosion on pipelines in both high pH and neutral pH carbonate/bicarbonate solutions. Corros. Sci. 2014, 85, 304–310. [Google Scholar] [CrossRef]
- Brenna, A.; Ormellese, M.; Lazzari, L. A proposal of AC corrosion mechanism in cathodic protection. NSTI Nanotech. 2011, 3, 553–556. [Google Scholar]
- Ibrahim, I.; Tribollet, B.; Takenouti, H.; Meyer, M. AC-induced corrosion of underground steel pipelines. Faradaic rectification under cathodic protection: I. Theoretical approach with negligible electrolyte resistance. J. Braz. Chem. 2015, 1, 196–208. [Google Scholar] [CrossRef]
- Marshakov, A.I.; Nenasheva, T.A.; Kasatkin, V.E.; Kasatkina, I.V. Effect of alternating current on dissolution rate of carbon steel in chloride electrolyte. II. Cathodic potentials. Prot. Metals Phys. Chem. Surf. 2018, 7, 1236–1245. [Google Scholar] [CrossRef]
- Buchler, M. Alternating current corrosion of cathodically protected pipelines: Discussion of the involved processes and their consequences on the critical interference values. Mater. Corros. 2012, 12, 1181–1187. [Google Scholar] [CrossRef]
- Huo, Y.; Tan, M.Y.; Forsyth, M. Visualizing dynamic passivation and localized corrosion processes occurring on buried steel surfaces under the effect of anodic transients. Electrochem. Commun. 2016, 66, 21–24. [Google Scholar] [CrossRef]
- Huo, Y.; Tan, M.Y. Measuring and understanding the critical duration and amplitude of anodic transients. Corros. Eng. Sci. Technol. 2018, 53, 65–72. [Google Scholar] [CrossRef]
- Huo, Y.; Tan, M.Y. Localized corrosion of cathodically protected pipeline steel under the effects of cyclic potential transients. Corros. Eng. Sci. Technol. 2018, 5, 348–354. [Google Scholar] [CrossRef]
- Liu, Z.Y.; Li, X.G.; Cheng, Y.F. Understand the occurrence of pitting corrosion of pipeline carbon steel under cathodic polarization. Electrochim. Acta 2012, 60, 259–263. [Google Scholar] [CrossRef]
- Marshakov, A.I.; Nenasheva, T.A. The formation of corrosion defects upon cathodic polarization of X70 grade pipe steel. Prot. Met. Phys. Chem. Surf. 2015, 7, 1122–1132. [Google Scholar] [CrossRef]
- Nenasheva, T.A.; Marshakov, A.I.; Kasatkina, I.V. The formation of local foci of corrosion of pipe steel under cyclic alternating polarization. Corros. Mater. Prot. 2015, 5, 9–17. (In Russian) [Google Scholar]
- Dai, M.; Liu, J.; Huang, F.; Zhang, Y.; Cheng, Y.F. Effect of cathodic protection potential fluctuations on pitting corrosion of X100 pipeline steel in acidic soil environment. Corros. Sci. 2018, 143, 428–437. [Google Scholar] [CrossRef]
- Parkins, R.N.; Blanchard, W.K.; Delanty, B.S. Transgranular stress corrosion cracking of high-pressure pipelines in contact with solutions of near neutral pH. Corrosion 1994, 5, 394–408. [Google Scholar] [CrossRef]
- Gladkikh, N.A.; Maleeva, M.A.; Maksaeva, L.B.; Petrunin, M.A. A study of the initial stages of local dissolution of carbon steel in chloride solution. Prot. Met. Phys. Chem. Surf. 2018, 7, 1260–1265. [Google Scholar] [CrossRef]
- Devanathan, M.A.V.; Stachurski, Z. The mechanism of hydrogen evolution on iron in acid solution by determination of permition rates. J. Electrochem. Soc. 1964, 3, 619–623. [Google Scholar] [CrossRef]
- Gladkikh, N.A.; Maleeva, M.A.; Maksaeva, L.B.; Petrunin, M.A.; Rybkina, A.A.; Yurasova, T.A.; Marshakov, A.I.; Zalavutdinov, R.K. Localized dissolution of carbon steel used for pipelines under constant cathodic polarization conditions. Initial stages of defect formation. Int. J. Corros. Scale Inhib. 2018, 4, 683–696. [Google Scholar]
- Marshakov, A.I.; Nenasheva, T.A. Kinetics of dissolution of hydrogenated carbon steel in electrolytes with pH close to neutral. Prot. Met. Phys. Chem. Surf. 2015, 6, 1018–1026. [Google Scholar]
- Rybkina, A.A.; Kasatkina, I.V.; Gladkikh, N.A.; Petrunin, M.A.; Marshakov, A.I. Experimental determination of local corrosion damage development rate on surface of pipe steels in model soil electrolytes. Corros. Mat. Prot. 2019, 3, 1–8. (In Russian) [Google Scholar]
- Marshakov, A.I.; Rybkina, A.A.; Maleeva, M.A.; Rybkin, A.A. The effect of atomic hydrogen on the kinetics of iron passivation in neutral solution. Prot. Met. Phys. Chem. Surf. 2014, 3, 345–351. [Google Scholar] [CrossRef]
- Marshakov, A.I.; Rybkina, A.A.; Maksaeva, L.B.; Petrunin, M.A.; Nazarov, A.P. A study of the initial stages of iron passivation in neutral solutions using the quartz crystal resonator technique. Prot. Met. Phys. Chem. Surf. 2016, 5, 936–946. [Google Scholar] [CrossRef]
- Cherry, B.W.; Gould, A.N. Pitting corrosion of nominally protected land–based pipelines. Mater. Perform. 1990, 4, 22–26. [Google Scholar]
- Marshakov, A.I.; Rybkina, A.A. Dissolution of iron and ionisation of hydrogen in borate buffer under cyclic pulse polarization. Int. J. Electrochem. Sci. 2019, 14, 9468–9481. [Google Scholar] [CrossRef]
- Morozova, N.B.; Vvedensky, A.V.; Beredina, I.P. The phase-boundary exchange and the non-steady-state diffusion of atomic hydrogen in Cu-Pd and Ag-Pd alloys. Part I. Analysis of the model. Prot. Met. Phys. Chem. Surf. 2014, 6, 699–704. [Google Scholar] [CrossRef]
- Zakroczymski, T. Adaptation of the electrochemical permeation technique for studying entry, transport and trapping of hydrogen in metals. Electrochim. Acta 2006, 11, 2261–2266. [Google Scholar] [CrossRef]
- Pyun, S.I.; Oriani, R.A. The permeation of hydrogen through the passivating films on iron and nickel. Corros. Sci. 1989, 5, 485–496. [Google Scholar] [CrossRef]
- Legrand, E.; Bouhattate, J.; Feaugas, X.; Garmestani, H. Computational analysis of geometrical factors affecting experimental data extracted from hydrogen permeation tests: II-consequences of trapping and an oxide layer. Int. J. Hydrogen Energy 2012, 37, 13574–13582. [Google Scholar] [CrossRef]
- Qin, Z.; Demko, B.; Noel, J.; Shoesmith, D.; King, F.; Worthingham, R.; Keith, K. Localized dissolution of mill scale-covered pipeline steel surfaces. Corrosion 2004, 10, 906–914. [Google Scholar] [CrossRef]
- Marshakov, I.K. Corrosion resistance and dezincing of brasses. Prot. Met. Phys. Chem. Surf. 2005, 3, 205–210. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
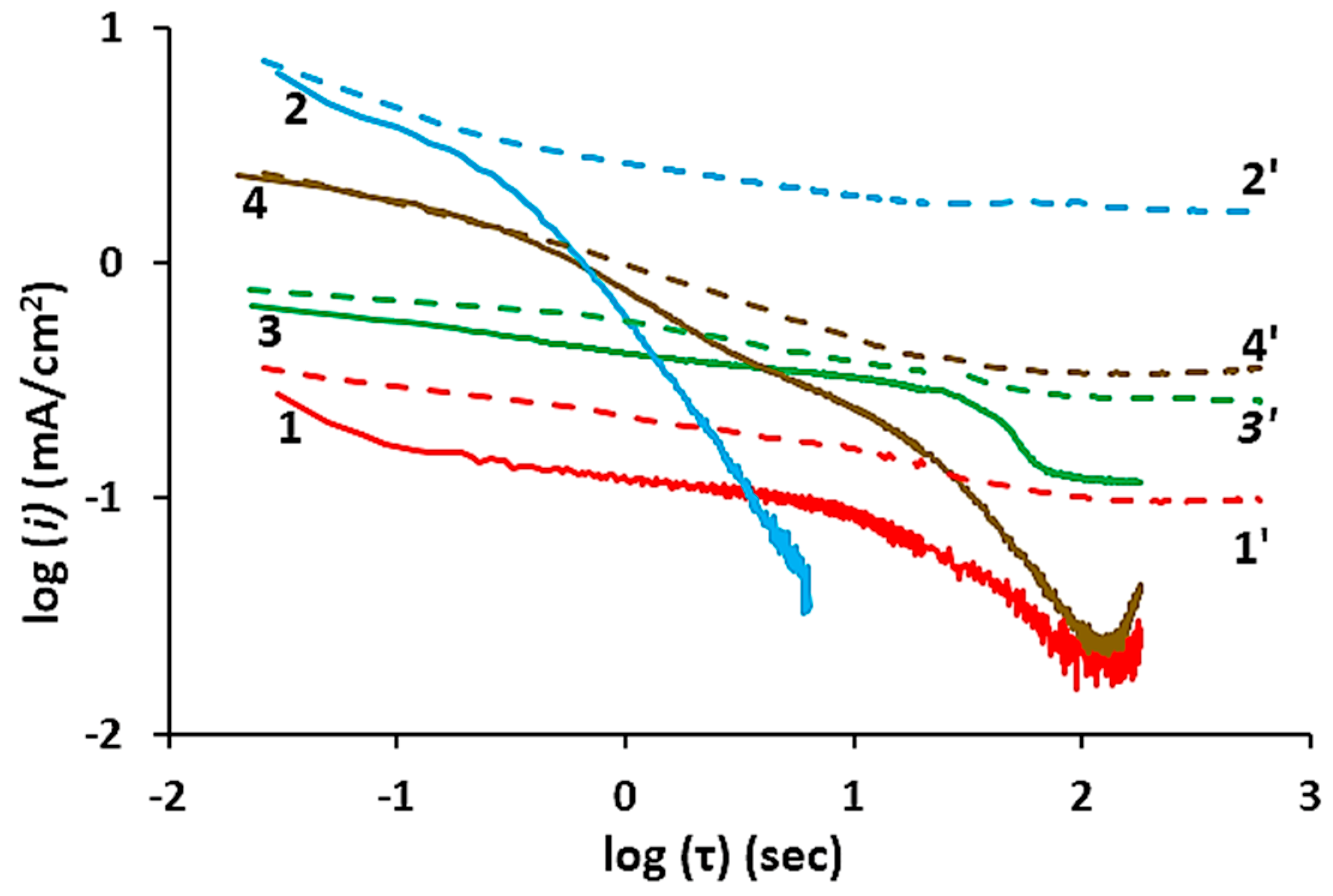{kind=link}
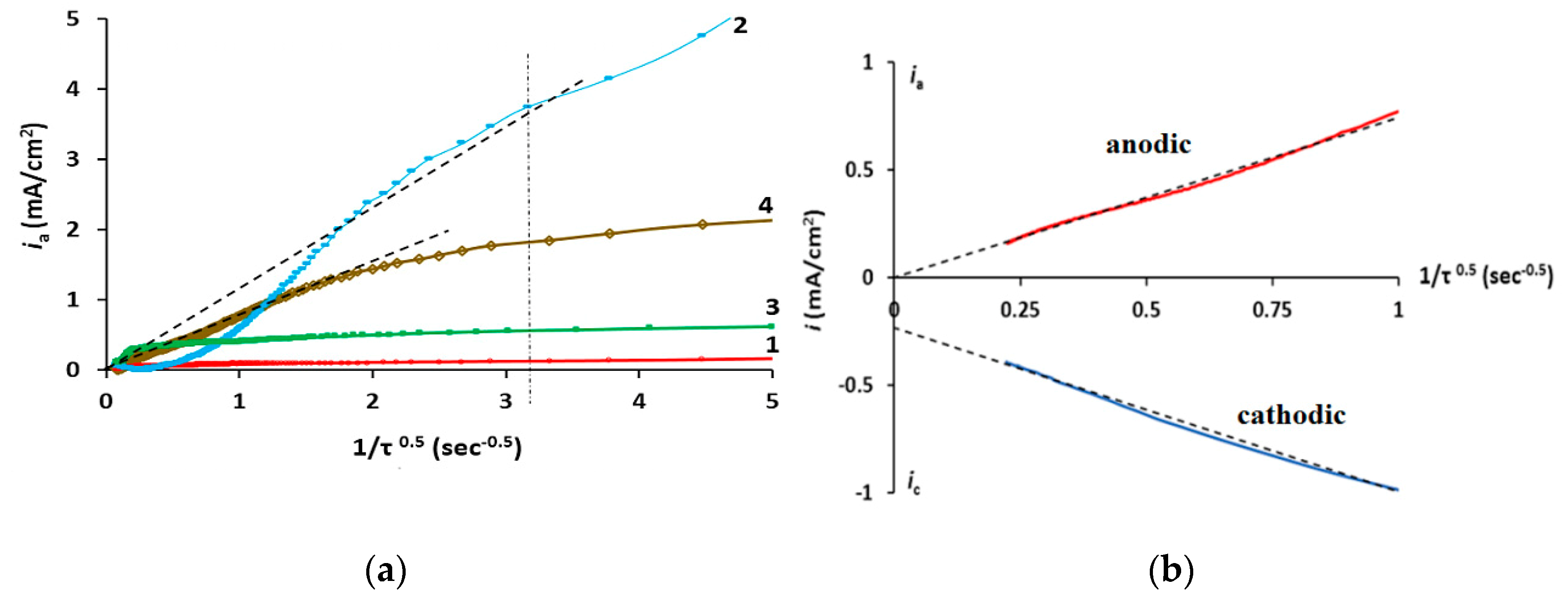{kind=link}
{kind=link}
| C | Si | Mn | P | S | Cr | Ni | Cu | Al | Ti |
|---|---|---|---|---|---|---|---|---|---|
| 0.115 | 0.34 | 1.63 | 0.021 | 0.003 | 0.04 | 0.02 | 0.007 | 0.030 | 0.07 |
| Electrolyte Composition | τc, min | Sp × 103, mm2/cm2 | ρ, pits/mm2 | d, µm | h, µm |
|---|---|---|---|---|---|
| BB | 10 | 0.006 | 2 | 2 | 1 |
| BB + NS4 | 0 | 0.73 | 22 | 6.5 | 3 |
| BB + NS4 | 10 | 1.13 | 32 | 6.7 | 4 |
| BB + NS4 | 60 | 1.66 | 30 | 8.4 | 4 |
| BB + NS4 + TU | 0 | 1.63 | 23 | 9.5 | 5 |
| BB + NS4 + TU | 10 | 4.72 | 68 | 9.4 | 7.5 |
| BB + NS4 + TU | 60 | 17.90 | 100 | 15.1 | 9 |
| 0.1 M NaCl | 10 | 3.56 | 60 | 8.7 | 10 |
| BB + 0.1 M NaCl | 10 | 2.72 | 60 | 7.6 | 6.5 |
| BB + 0.6 M NaCl | 10 | 5.17 | 39 | 13 | 7.5 |
| BB + 0.1 M NaHCO3 | 10 | 0.84 | 17.5 | 7.8 | 2 |
| BB + 0.1 M NaHCO3 + TU | 10 | 2.27 | 43 | 8.2 | 3 |
| BB + NS4 + TU + IFKhAN-29 | 60 | 1.76 | 22 | 10.1 | 4 |
| Electrolyte Composition | ip, A/cm2 | ia,st, mA/cm2 | i0, mA/cm2 |
|---|---|---|---|
| BB | 6 | 0.020 | 0.17 |
| BB + NS4 | 10 | 0.020 | 0.20 |
| BB + NS4 + TU | 90 | 0.116 | 0.57 |
| 0.1 M NaCl | 14.5 | 0.041 | 1.85 |
| BB + 0.1 M NaCl | 18 | 0.100 | 1.27 |
| BB +0.6 M NaCl | 38 | 0.082 | 3.8 |
| BB + 0.1 M NaHCO3 | 18 | 0.067 | 0.44 |
| BB + 0.1 M NaHCO3 + TU | 27.6 | 0.113 | 1.05 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rybkina, A.; Gladkikh, N.; Marshakov, A.; Petrunin, M.; Nazarov, A. Effect of Sign-Alternating Cyclic Polarisation and Hydrogen Uptake on the Localised Corrosion of X70 Pipeline Steel in Near-Neutral Solutions. Metals 2020, 10, 245. https://doi.org/10.3390/met10020245
Rybkina A, Gladkikh N, Marshakov A, Petrunin M, Nazarov A. Effect of Sign-Alternating Cyclic Polarisation and Hydrogen Uptake on the Localised Corrosion of X70 Pipeline Steel in Near-Neutral Solutions. Metals. 2020; 10(2):245. https://doi.org/10.3390/met10020245
Chicago/Turabian StyleRybkina, Alevtina, Natalia Gladkikh, Andrey Marshakov, Maxim Petrunin, and Andrei Nazarov. 2020. "Effect of Sign-Alternating Cyclic Polarisation and Hydrogen Uptake on the Localised Corrosion of X70 Pipeline Steel in Near-Neutral Solutions" Metals 10, no. 2: 245. https://doi.org/10.3390/met10020245