

Chromosomal-Level Reference Genome for the Chinese Endemic Pygmy Grasshopper, Zhengitettix transpicula, Sheds Light on Tetrigidae Evolution and Advancing Conservation Efforts


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Specimen Acquisition
2.2. Genomic and Transcriptomic Sequencing
2.3. Sequencing Data Quality Control
2.4. Estimating Genomic Size and Heterozygosity
2.5. Genomic Assembly and Evaluation of Quality
2.6. Comparative Genomic Analysis
3. Results
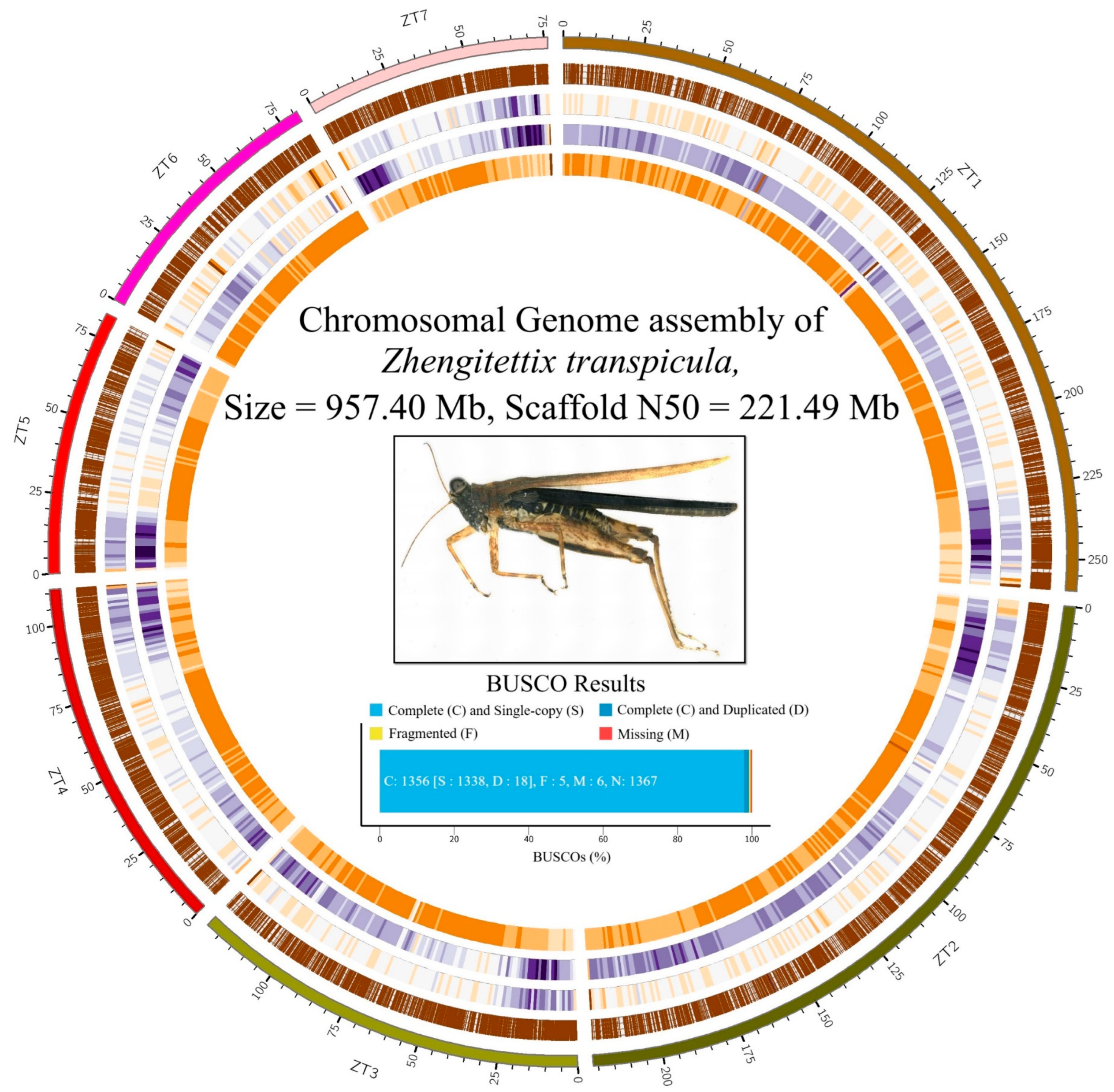
3.1. Genome Assembly
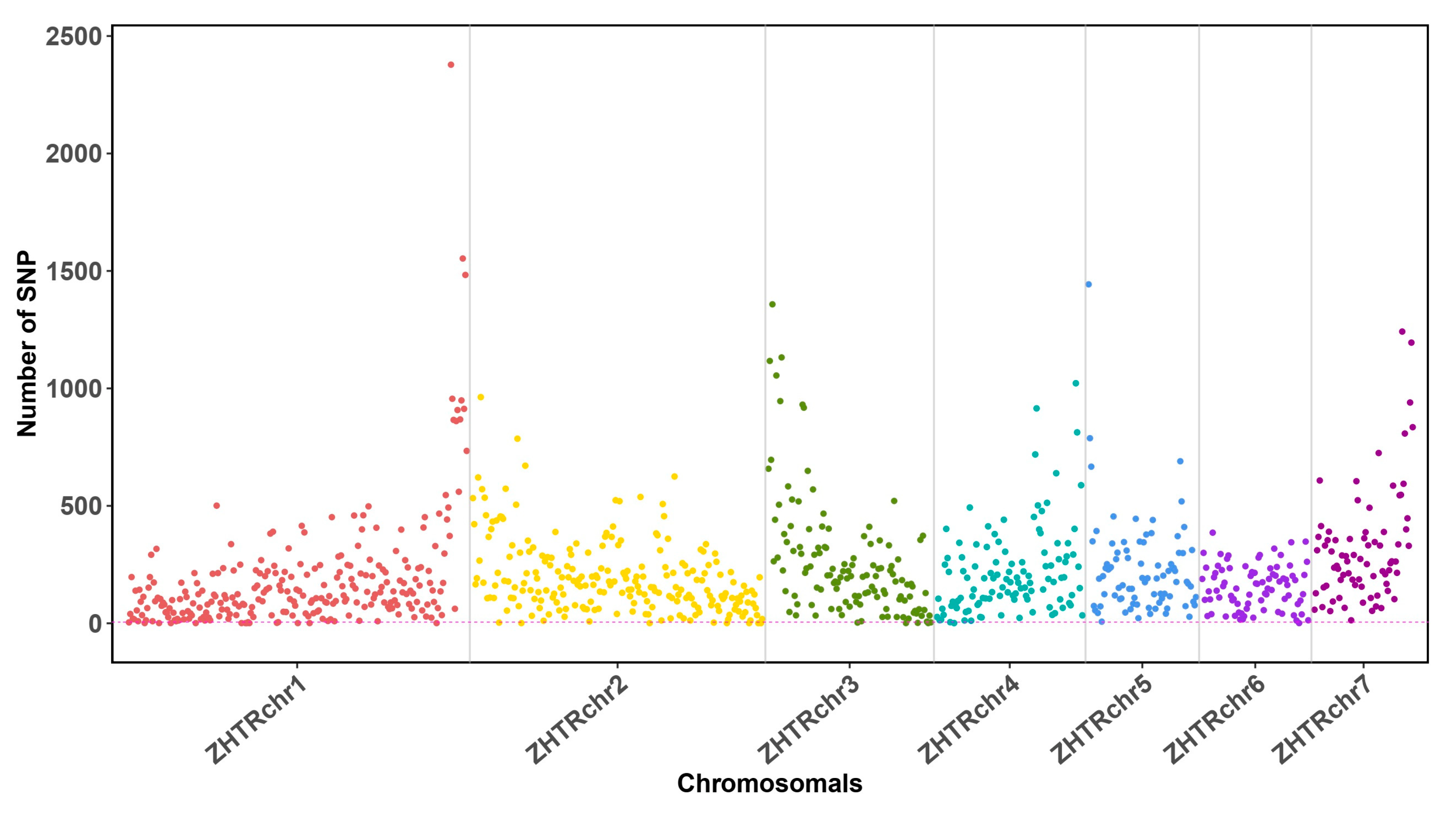
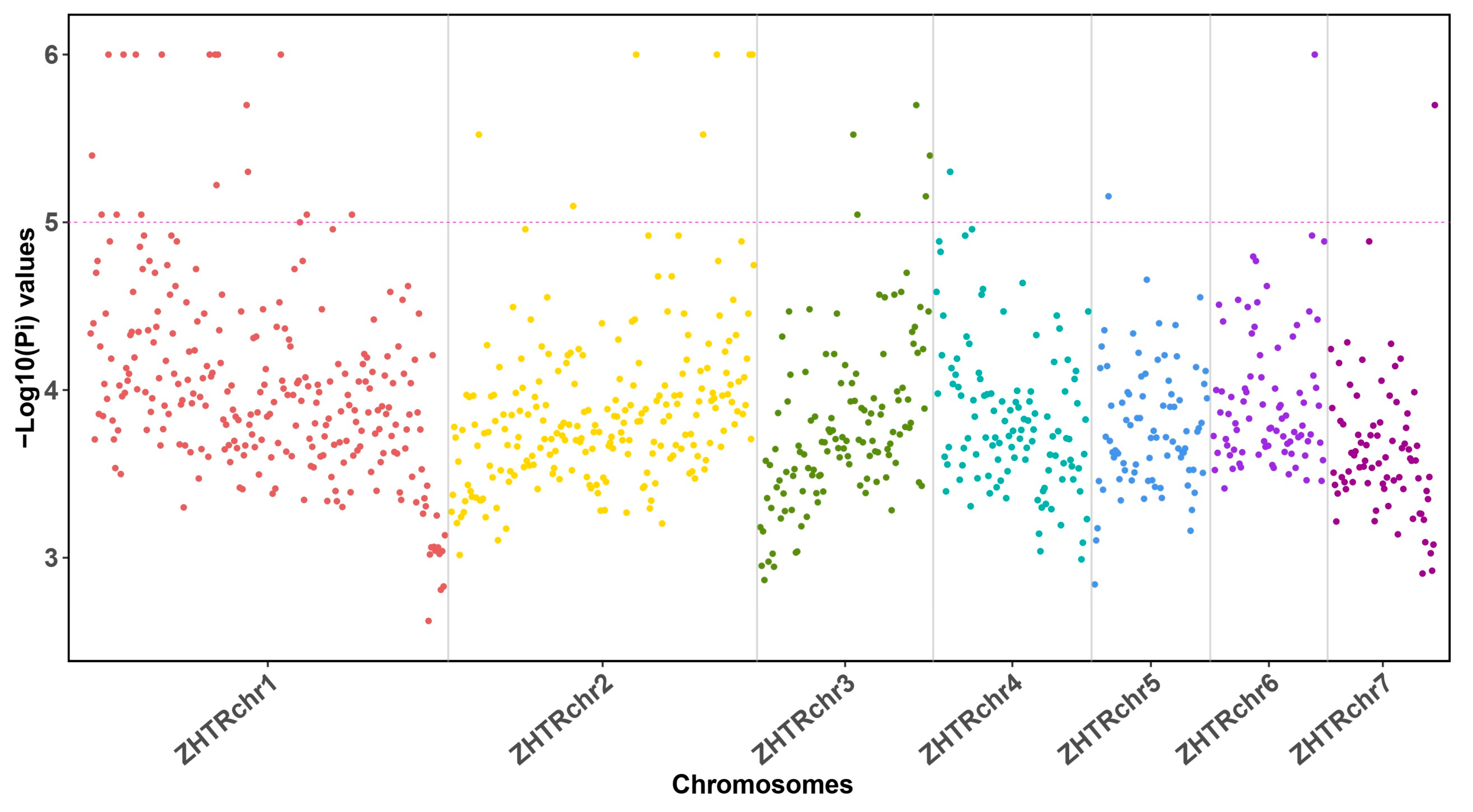
3.2. Low Genetic Heterozygosity Suggests a Small Effective Population Size in Z. transpicula
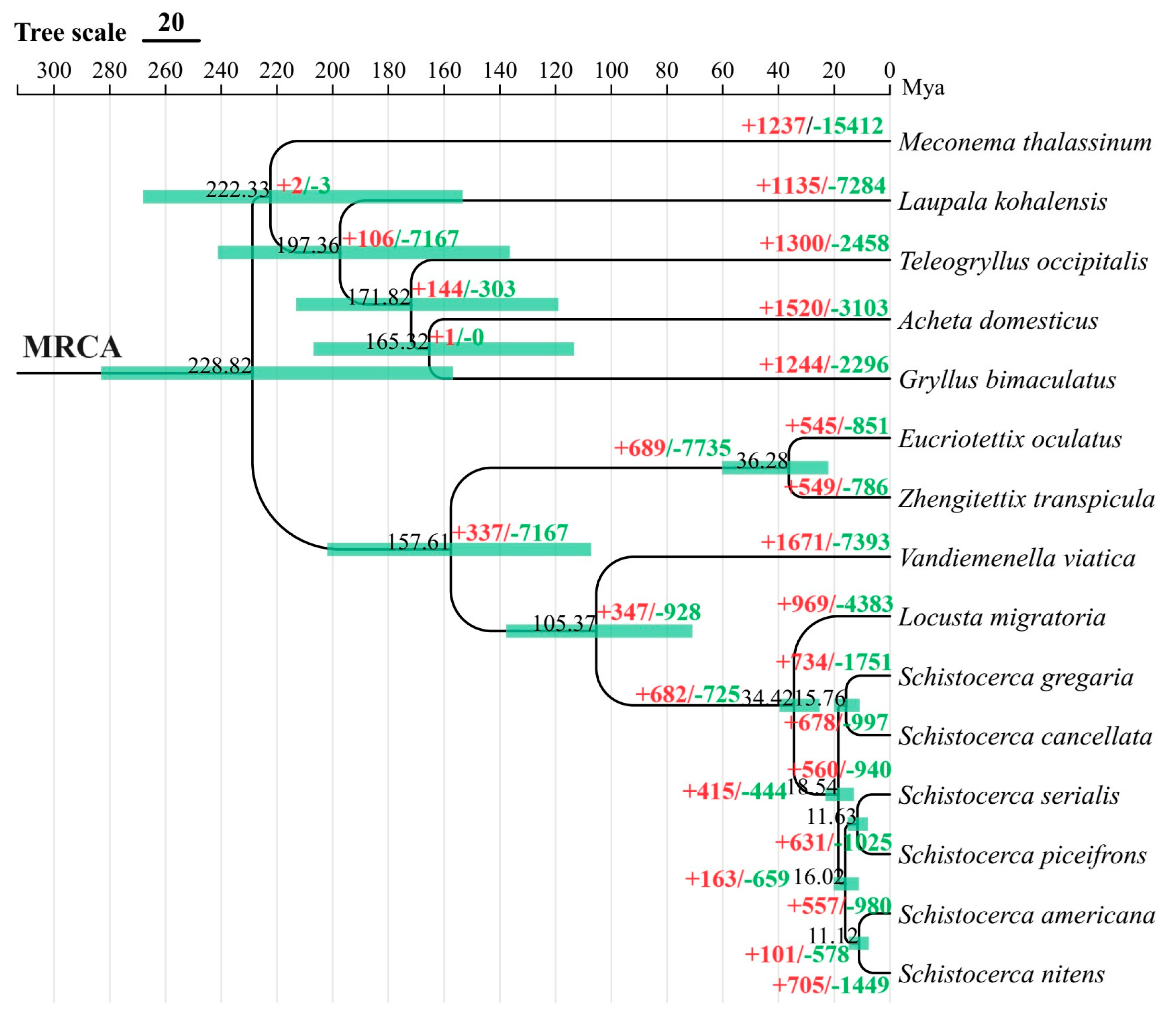
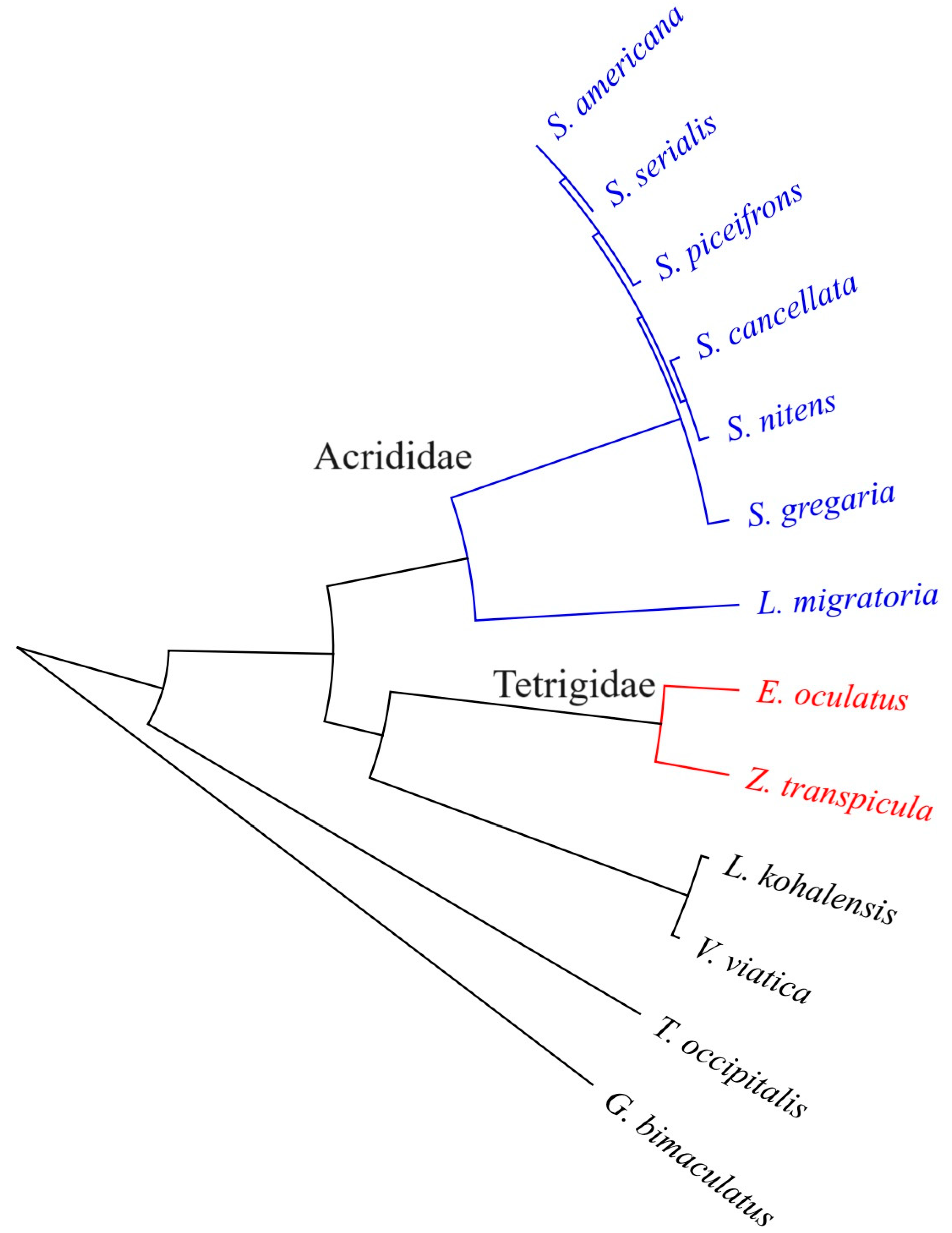
3.3. Divergence Time Estimation and Gene Family Expansion/Construction Analysis
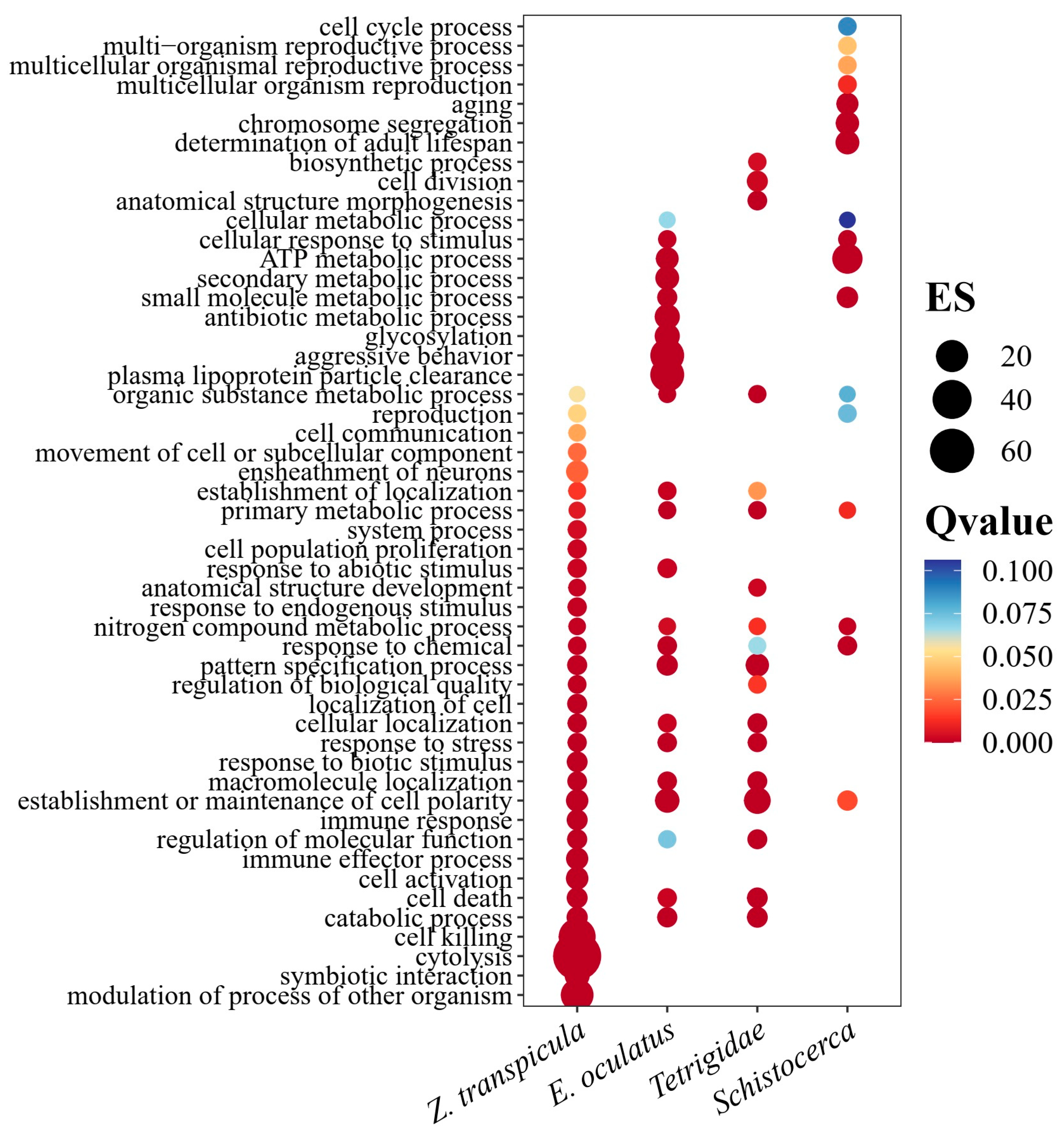
3.4. Divergent Evolution of the CYP305m2 Gene in Tetrigidae and Acrididae
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Storozhenko, S.Y.; Dawwrueng, P. New and little-known pygmy grasshoppers (Orthoptera: Tetrigidae) from Thailand. Zootaxa 2015, 4052, 527–554. [Google Scholar] [CrossRef]
- Deng, W.A. New genus and new species of leaf-mimic pygmy grasshoppers from China (Orthoptera: Tetrigidae: Cladonotinae). Zootaxa 2021, 4995, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Forsman, A. On the role of sex differences for evolution in heterogeneous and changing fitness landscapes: Insights from pygmy grasshoppers. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 2018, 373, 20170429. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Liu, F.; Lu, H.; Huang, Y. Characterization and analysis of a de novo transcriptome from the pygmy grasshopper Tetrix japonica. Mol. Ecol. Resour. 2017, 17, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Kuřavová, K.; Šipoš, J.; Kočárek, P. Energy balance of food in a detrito-bryophagous groundhopper (Orthoptera: Tetrigidae). PeerJ 2020, 8, e9603. [Google Scholar] [CrossRef] [PubMed]
- Wennersten, L.; Johansson, J.; Karpestam, E.; Forsman, A. Higher establishment success in more diverse groups of pygmy grasshoppers under seminatural conditions. Ecology 2012, 93, 2519–2525. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.Q.; Shi, J.P.; Yin, Z. Discovery of a new species and key to all known species of the genus Aalatettix Zheng & Mao (Orthoptera, Tetrigoidea, Tetrigidae) from China. Zootaxa 2016, 4097, 143–146. [Google Scholar] [CrossRef]
- Forsman, A.; Karlsson, M.; Wennersten, L.; Johansson, J.; Karpestam, E. Rapid evolution of fire melanism in replicated populations of pygmy grasshoppers. Evol. Int. J. Org. Evol. 2011, 65, 2530–2540. [Google Scholar] [CrossRef]
- Bastow, J.L.; Sabo, J.L.; Finlay, J.C.; Power, M.E. A basal aquatic-terrestrial trophic link in rivers: Algal subsidies via shore-dwelling grasshoppers. Oecologia 2002, 131, 261–268. [Google Scholar] [CrossRef]
- Chen, Y.Z.; Deng, W.A. Two new species of the genus Zhengitettix (Orthoptera: Tetrigidae: Scelimeninae) from China. Zootaxa 2022, 5154, 71–80. [Google Scholar] [CrossRef]
- Chen, Y.Z.; Deng, W.A.; Wang, J.M.; Lin, L.L.; Zhou, S.Y. Phylogenetic relationships of Scelimeninae genera (Orthoptera: Tetrigoidea) based on COI, 16S rRNA and 18S rRNA gene sequences. Zootaxa 2018, 4482, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Singla, D.; Yadav, I.S. GAAP: A GUI-based Genome Assembly and Annotation Package. Curr. Genom. 2022, 23, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Hesse, U. K-Mer-Based Genome Size Estimation in Theory and Practice. Methods Mol. Biol. 2023, 2672, 79–113. [Google Scholar] [CrossRef] [PubMed]
- Ranallo-Benavidez, T.R.; Jaron, K.S.; Schatz, M.C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020, 11, 1432. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Song, L.; Wang, X.; Cheng, H.; Wang, C.; Meyer, C.A.; Liu, T.; Tang, M.; Aluru, S.; Yue, F.; et al. Fast alignment and preprocessing of chromatin profiles with Chromap. Nat. Commun. 2021, 12, 6566. [Google Scholar] [CrossRef]
- Zhou, C.; McCarthy, S.A.; Durbin, R. YaHS: Yet another Hi-C scaffolding tool. Bioinformatics 2023, 39, btac808. [Google Scholar] [CrossRef]
- Rodriguez, M.; Makałowski, W. Software evaluation for de novo detection of transposons. Mob. DNA 2022, 13, 14. [Google Scholar] [CrossRef]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009, 25, 4.10.1–4.10.14. [Google Scholar] [CrossRef]
- Campbell, M.S.; Holt, C.; Moore, B.; Yandell, M. Genome Annotation and Curation Using MAKER and MAKER-P. Curr. Protoc. Bioinform. 2014, 48, 4.11.11–14.11.39. [Google Scholar] [CrossRef]
- Guo, H.; Huang, C.; Jiang, L.; Cheng, T.; Feng, T.; Xia, Q. Transcriptome analysis of the response of silkworm to drastic changes in ambient temperature. Appl. Microbiol. Biotechnol. 2018, 102, 10161–10170. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods Mol. Biol. 2019, 1962, 227–245. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Yang, Z. PAMLX: A graphical user interface for PAML. Mol. Biol. Evol. 2013, 30, 2723–2724. [Google Scholar] [CrossRef] [PubMed]
- De Bie, T.; Cristianini, N.; Demuth, J.P.; Hahn, M.W. CAFE: A computational tool for the study of gene family evolution. Bioinformatics 2006, 22, 1269–1271. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Westbury, M.V.; Hartmann, S.; Barlow, A.; Wiesel, I.; Leo, V.; Welch, R.; Parker, D.M.; Sicks, F.; Ludwig, A.; Dalén, L.; et al. Extended and Continuous Decline in Effective Population Size Results in Low Genomic Diversity in the World’s Rarest Hyena Species, the Brown Hyena. Mol. Biol. Evol. 2018, 35, 1225–1237. [Google Scholar] [CrossRef]
- Gu, J.J.; Yue, Y.; Shi, F.; Tian, H.; Ren, D. First Jurassic grasshopper (Insecta, Caelifera) from China. Zootaxa 2016, 4169, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Misof, B.; Liu, S.; Meusemann, K.; Peters, R.S.; Donath, A.; Mayer, C.; Frandsen, P.B.; Ware, J.; Flouri, T.; Beutel, R.G.; et al. Phylogenomics resolves the timing and pattern of insect evolution. Science 2014, 346, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Forni, G.; Plazzi, F.; Cussigh, A.; Conle, O.; Hennemann, F.; Luchetti, A.; Mantovani, B. Phylomitogenomics provides new perspectives on the Euphasmatodea radiation (Insecta: Phasmatodea). Mol. Phylogenet. Evol. 2021, 155, 106983. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Cassau, S.; Krieger, J.; Guo, X.; Knaden, M.; Kang, L.; Hansson, B.S. A chemical defense deters cannibalism in migratory locusts. Science 2023, 380, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Shao, W.; Cao, M.; Ge, J.; Yang, P.; Chen, L.; Wang, X.; Kang, L. Phenylacetonitrile in locusts facilitates an antipredator defense by acting as an olfactory aposematic signal and cyanide precursor. Sci. Adv. 2019, 5, eaav5495. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Fang, X.; Yang, P.; Jiang, X.; Jiang, F.; Zhao, D.; Li, B.; Cui, F.; Wei, J.; Ma, C.; et al. The locust genome provides insight into swarm formation and long-distance flight. Nat. Commun. 2014, 5, 2957. [Google Scholar] [CrossRef]
- Verlinden, H.; Sterck, L.; Li, J.; Li, Z.; Yssel, A.; Gansemans, Y.; Verdonck, R.; Holtof, M.; Song, H.; Behmer, S.T.; et al. First draft genome assembly of the desert locust, Schistocerca gregaria. F1000Research 2020, 9, 775. [Google Scholar] [CrossRef]
- Guohui, Y.; Benyong, M.; Jishan, X.J.J.o.D.U. A Comparative Study on the Karyotype and C-band of Two Tetrix Species from Cangshan Mountain Region. J. Dali Univ. 2009, 8, 70–72. [Google Scholar]
- Mo, R.; Yao, G.; Wang, G.; Li, W. One new species and one new Chinese record of Neoperla (Plecoptera: Perlidae) from Mount Shiwandashan of the Guangxi Zhuang Autonomous Region of southern China. Zootaxa 2019, 4652, 497–506. [Google Scholar] [CrossRef]
- Chen, W.; Liao, X.; Zhou, S.; Mo, Y.; Huang, Y. Rediscovery of Rhacophorus yaoshanensis and Theloderma kwangsiensis at their type localities after five decades. Zootaxa 2018, 4379, 484–496. [Google Scholar] [CrossRef]
- Bush, S.E.; Duszynski, D.W.; Nickol, B.B. Acanthocephala from amphibians in China with the description of a new species of Pseudoacanthocephalus (Echinorhynchida). J. Parasitol. 2009, 95, 1440–1445. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Ying, X.; Deng, W.; Rong, W.; Li, X. Mitochondrial genomes of eight Scelimeninae species (Orthoptera) and their phylogenetic implications within Tetrigoidea. PeerJ 2021, 9, e10523. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Ma, C.; Kang, L. Community change and population outbreak of grasshoppers driven by climate change. Curr. Opin. Insect Sci. 2024, 61, 101154. [Google Scholar] [CrossRef]
- Liu, Y.; Li, X.; Lin, L. Transcriptome of the pygmy grasshopper Formosatettix qinlingensis (Orthoptera: Tetrigidae). PeerJ 2023, 11, e15123. [Google Scholar] [CrossRef] [PubMed]
- Tinnert, J.; Hellgren, O.; Lindberg, J.; Koch-Schmidt, P.; Forsman, A. Population genetic structure, differentiation, and diversity in Tetrix subulata pygmy grasshoppers: Roles of population size and immigration. Ecol. Evol. 2016, 6, 7831–7846. [Google Scholar] [CrossRef]
- Couzin, I.D.; Couzin-Fuchs, E. The chemical ecology of locust cannibalism. Science 2023, 380, 454–455. [Google Scholar] [CrossRef]
- Ernst, U.R.; Van Hiel, M.B.; Depuydt, G.; Boerjan, B.; De Loof, A.; Schoofs, L. Epigenetics and locust life phase transitions. J. Exp. Biol. 2015, 218, 88–99. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guan, D.-L.; Chen, Y.-Z.; Qin, Y.-C.; Li, X.-D.; Deng, W.-A. Chromosomal-Level Reference Genome for the Chinese Endemic Pygmy Grasshopper, Zhengitettix transpicula, Sheds Light on Tetrigidae Evolution and Advancing Conservation Efforts. Insects 2024, 15, 223. https://doi.org/10.3390/insects15040223
Guan D-L, Chen Y-Z, Qin Y-C, Li X-D, Deng W-A. Chromosomal-Level Reference Genome for the Chinese Endemic Pygmy Grasshopper, Zhengitettix transpicula, Sheds Light on Tetrigidae Evolution and Advancing Conservation Efforts. Insects. 2024; 15(4):223. https://doi.org/10.3390/insects15040223
Chicago/Turabian StyleGuan, De-Long, Ya-Zhen Chen, Ying-Can Qin, Xiao-Dong Li, and Wei-An Deng. 2024. "Chromosomal-Level Reference Genome for the Chinese Endemic Pygmy Grasshopper, Zhengitettix transpicula, Sheds Light on Tetrigidae Evolution and Advancing Conservation Efforts" Insects 15, no. 4: 223. https://doi.org/10.3390/insects15040223