
Interaction Analysis of lncRNA and mRNA Based on the Full-Length Transcriptome of the Nymph-to-Adult Developmental Transition of Sogatella furcifera

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Insect Rearing, Sample Collection, and RNA Extraction
2.2. Library Preparation and Sequencing Data Analysis
2.3. Identification of lncRNAs
2.4. Analysis of Differentially Expressed lncRNAs and Prediction of Target Genes
2.5. Expression Dynamic Analysis of lncRNAs and mRNAs
2.6. Interaction Analysis between lncRNAs and mRNAs
2.7. RT–qPCR Analysis
2.8. Statistical Analysis
3. Results
3.1. Identification and Characterization of lncRNAs in S. furcifera
3.2. Analysis of Differentially Expressed lncRNAs and mRNAs
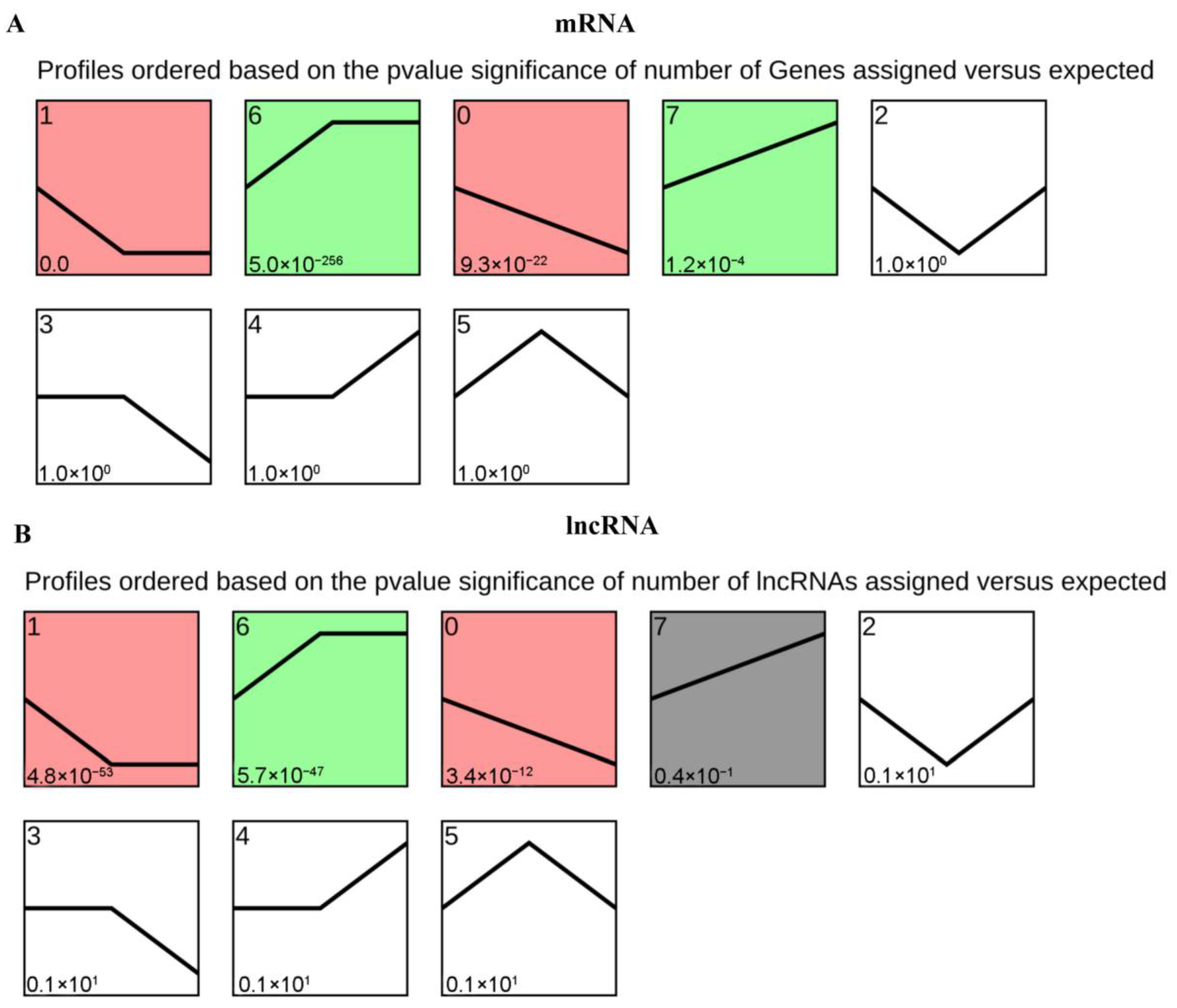
3.3. Analysis of lncRNA and mRNA Expression Patterns
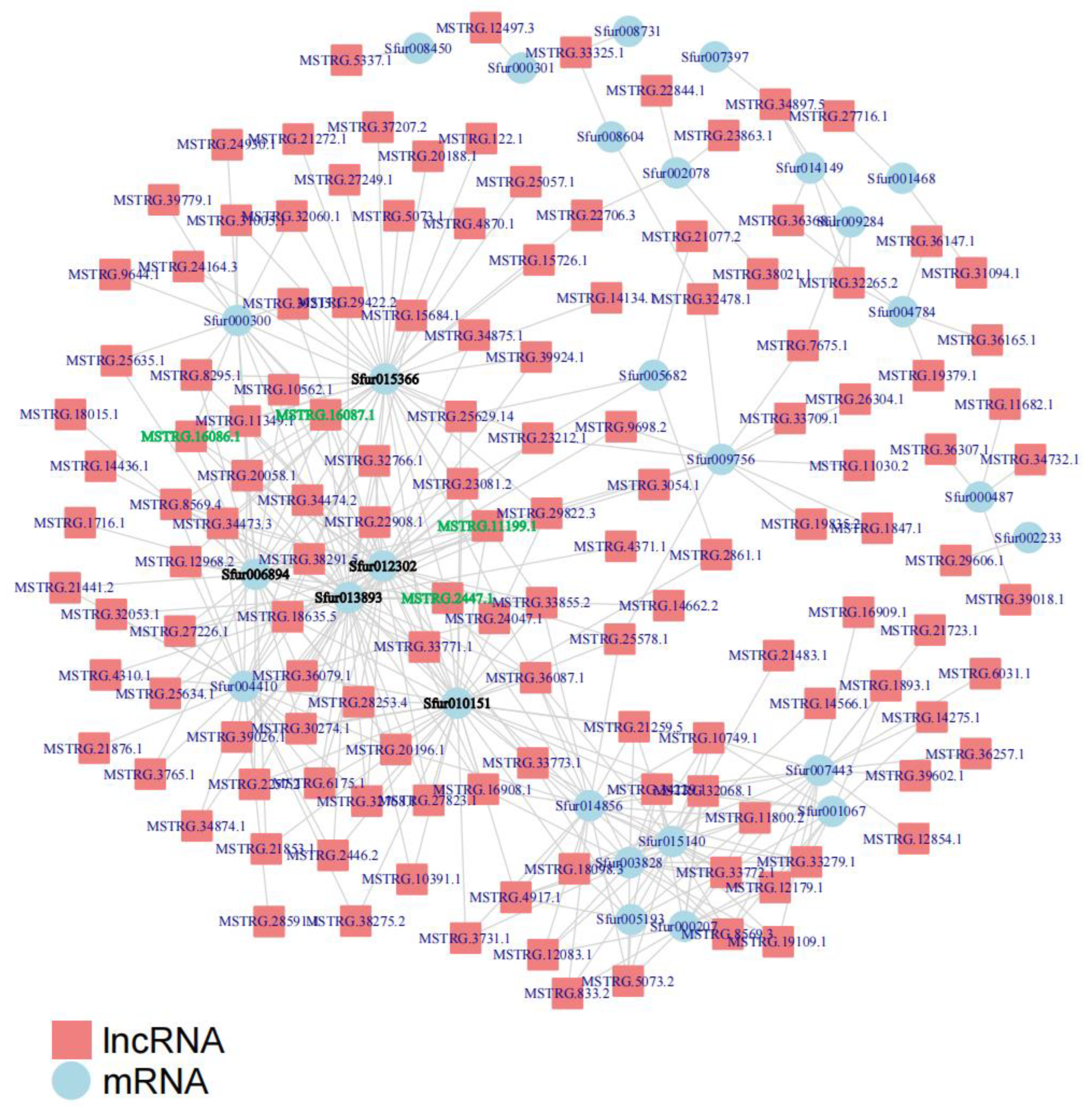
3.4. Analysis of the Interaction between Differentially Expressed lncRNAs and mRNAs
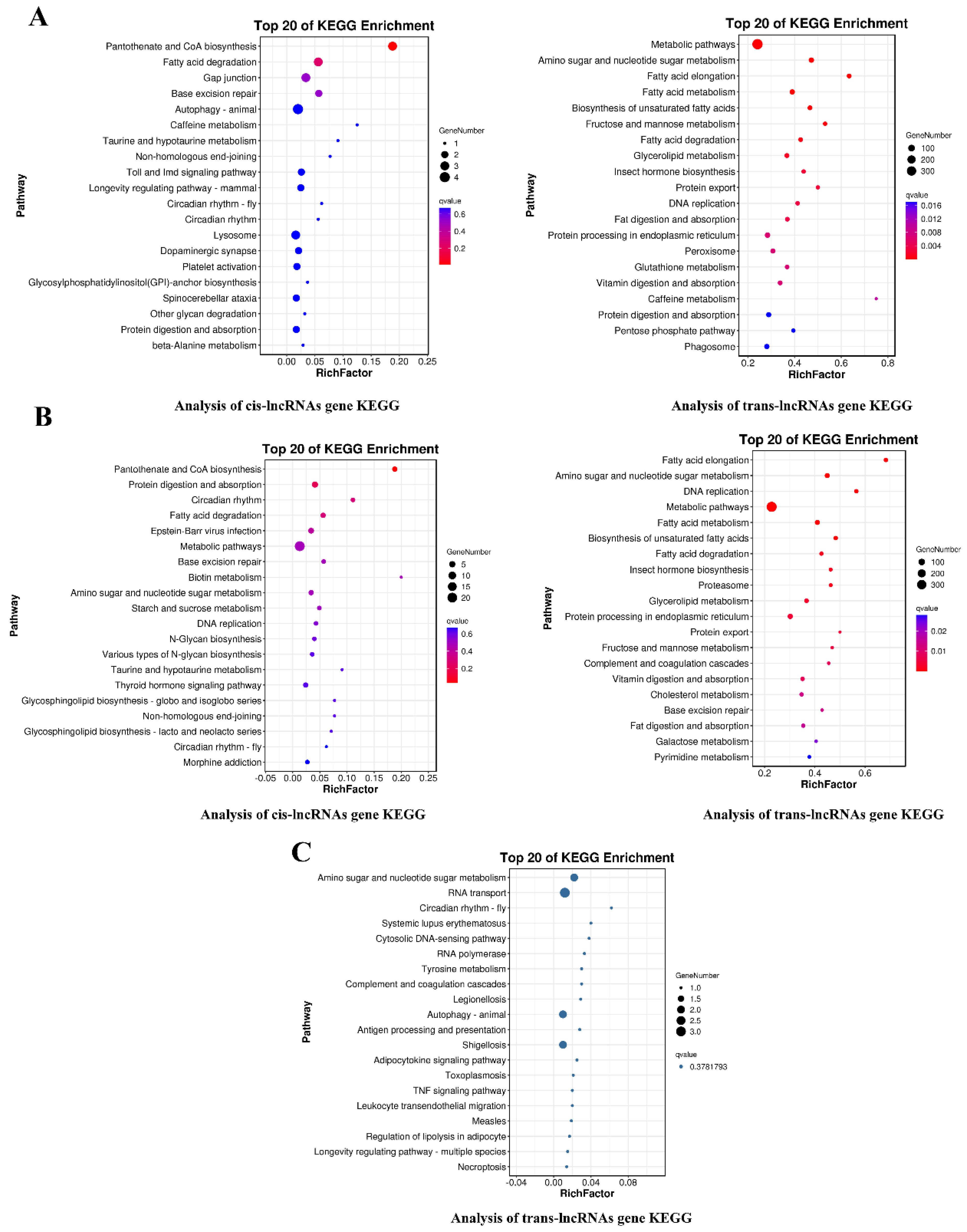
3.5. Prediction and Analysis of the Target Genes of Differentially Expressed lncRNAs
3.6. Verification of Sequencing Results
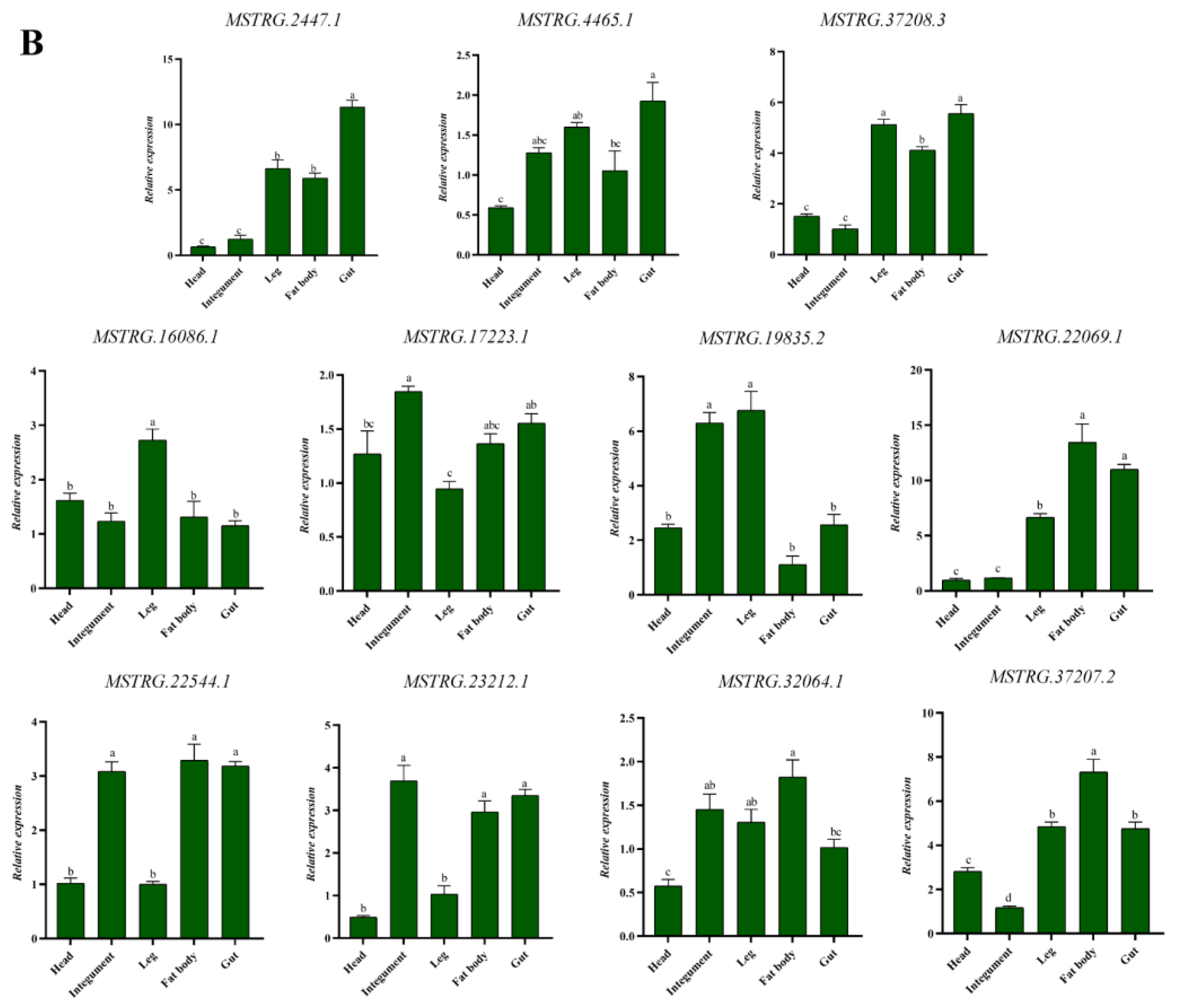
3.7. Spatial and Temporal Expression Patterns of Differentially Expressed lncRNAs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- St Laurent, G.; Wahlestedt, C.; Kapranov, P. The landscape of long noncoding RNA classification. Trends Genet. 2015, 31, 239–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.Z.; Xiao, H.M.; He, K.; Li, F. Progress and prospects of noncoding RNAs in insects. J. Integr. Agric. 2019, 18, 729–747. [Google Scholar] [CrossRef] [Green Version]
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermüller, J.; Hofacker, I.L.; et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 2007, 316, 1484–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdmann, V.A.; Szymanski, M.; Hochberg, A.; Groot, N.D.; Barciszewski, J. Non-coding, mRNA-like RNAs database Y2K. Nucleic Acids Res. 2000, 28, 197–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Okada, T.; Fukushima, T.; Tsudzuki, T.; Sugiura, M.; Yukawa, Y. A novel hypoxic stress-responsive long non-coding RNA transcribed by RNA polymerase III in Arabidopsis. RNA Biol. 2012, 9, 302–313. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Bajic, V.B.; Zhang, Z. On the classification of long non-coding RNAs. RNA Biol. 2013, 10, 925–933. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell. 2011, 43, 904–914. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.H.; Abdelmohsen, K.; Gorospe, M. Posttranscriptional gene regulation by long noncoding RNA. J. Mol. Biol. 2013, 425, 3723–3730. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Lu, X.; Yuan, L. lncRNA: A link between RNA and cancer. Biochim. Biophys. Acta. 2014, 1839, 1097–1109. [Google Scholar] [CrossRef]
- Yao, R.W.; Wang, Y.; Chen, L.L. Cellular functions of long noncoding RNAs. Nat. Cell Biol. 2019, 21, 542–551. [Google Scholar] [CrossRef]
- Azlan, A.; Obeidat, S.M.; Yunus, M.A.; Azzam, G. Systematic identification and characterization of Aedes aegypti long noncoding RNAs (lncRNAs). Sci. Rep. 2019, 9, 12147. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Wang, Y.W.; Lu, Y.Y.; Li, B.; Chen, K.P.; Li, C.J. Genome-wide identification and characterization of long non-coding RNAs in Tribolium castaneum. Insect Sci. 2021, 28, 1262–1276. [Google Scholar] [CrossRef] [PubMed]
- Humann, F.C.; Tiberio, G.J.; Hartfelder, K. Sequence and expression characteristics of long noncoding RNAs in honey bee caste development–potential novel regulators for transgressive ovary size. PLoS ONE 2013, 8, e78915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, H.; Yuan, Z.; Guo, D.; Hou, B.; Yin, C.; Zhang, W.; Li, F. Genome-wide identification of long noncoding RNA genes and their potential association with fecundity and virulence in rice brown planthopper, Nilaparvata lugens. BMC Genom. 2015, 16, 749. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Li, W.L.; Zeng, H.X.; Shi, Y.; Liao, X.L. Systematic identification and functional analysis of long noncoding RNAs involved in indoxacarb resistance in Spodoptera litura. Insect Sci. 2022, 29, 1721–1736. [Google Scholar] [CrossRef] [PubMed]
- Valanne, S.; Salminen, T.S.; Järvelä-Stölting, M.; Vesala, L.; Rämet, M. Immune-inducible non-coding RNA molecule lincRNA-IBIN connects immunity and metabolism in Drosophila melanogaster. PLoS Pathog. 2019, 15, e1007504. [Google Scholar]
- Meng, L.W.; Yuan, G.R.; Chen, M.L.; Dou, W.; Jing, T.X.; Zheng, L.S.; Peng, M.L.; Bai, W.J.; Wang, J.J. Genome-wide identification of long non-coding RNAs (lncRNAs) associated with Malathion resistance in Bactrocera dorsalis. Pest. Manag. Sci. 2021, 77, 2292–2301. [Google Scholar] [CrossRef]
- Liu, F.; Guo, D.; Yuan, Z.; Chen, C.; Xiao, H.M. Genome-wide identification of long non-coding RNA genes and their association with insecticide resistance and metamorphosis in diamondback moth, Plutella xylostella. Sci. Rep. 2017, 7, 15870. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Yu, Y.; Kang, K.; Zhang, D.W. SMRT sequencing of the full-length transcriptome of the white-backed planthopper Sogatella furcifera. PeerJ. 2020, 8, e9320. [Google Scholar] [CrossRef]
- Chang, Z.X.; Ajayi, O.E.; Guo, D.Y.; Wu, Q.F. Genome-wide characterization and developmental expression profiling of long non-coding RNAs in Sogatella furcifera. Insect Sci. 2020, 27, 987–997. [Google Scholar] [CrossRef]
- Zhou, G.H.; Wen, J.J.; Cai, D.J.; Li, P.; Xu, D.L.; Zhang, S.G. Southern rice black-streaked dwarf virus: A new proposed Fijivirus species in the family Reoviridae. Chin. Sci. Bull. 2008, 53, 3677–3685. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, W.; Xu, L.; Hua, H.; Chen, M.Y.; Guo, M.J.; He, K.; Zhao, J.; Li, F. Chromosomal-level genomes of three rice planthoppers provide new insights into sex chromosome evolution. Mol. Ecol. Resour. 2021, 21, 226–237. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, stringTie and ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Kang, Y.J.; Yang, D.C.; Kong, L.; Kong, L.; Meng, Y.Q.; Wei, L.P.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Li, G.; Liu, X.Y.; Smagghe, G.; Niu, J.Z.; Wang, J.J. Genome-wide characterization and identification of long non-coding RNAs during the molting process of a Spider Mite, Panonychus citri. Int. J. Mol. Sci. 2021, 22, 6909. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhang, Y.; Zhang, X.; Jia, S.; Chen, S.; Kang, L. Genome-wide identification and developmental expression profiling of long noncoding RNAs during Drosophila metamorphosis. Sci. Rep. 2016, 6, 23330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.B.; Zhou, C.; Long, G.Y.; Yang, H.; Chen, C.; Jin, D.C. Characterization and functional analysis of chitinase family genes involved in nymph–adult transition of Sogatella furcifera. Insect Sci. 2021, 28, 901–916. [Google Scholar] [CrossRef]
- Yang, X.B.; Zhou, C.; Gong, M.F.; Yang, H.; Long, G.Y.; Jin, D.C. Identification and RNAi-based functional analysis of four chitin deacetylase genes in Sogatella furcifera (Hemiptera: Delphacidae). Insect Sci. 2021, 21, 9. [Google Scholar] [CrossRef]
- Wang, Z.; Zhou, C.; Long, G.Y.; Yang, H.; Jin, D.C. Sublethal effects of buprofezin on development, reproduction, and chitin synthase 1 gene (SfCHS1) expression in the white-backed planthopper, Sogatella furcifera (Hemiptera: Delphacidae). Asia-Pac. Entomol. 2018, 21, 585–591. [Google Scholar] [CrossRef]
- Pan, P.L.; Ye, Y.X.; Lou, Y.H.; Lu, J.B.; Cheng, C.; Shen, Y.; Moussian, B.; Zhang, C.X. A comprehensive omics analysis and functional survey of cuticular proteins in the brown planthopper. Proc. Natl. Acad. Sci. USA 2018, 115, 5175–5180. [Google Scholar] [CrossRef] [Green Version]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, Z.; Yang, X.; Yang, H.; Dai, R.; Zeng, Q.; Jin, D. Interaction Analysis of lncRNA and mRNA Based on the Full-Length Transcriptome of the Nymph-to-Adult Developmental Transition of Sogatella furcifera. Insects 2023, 14, 308. https://doi.org/10.3390/insects14040308
Jia Z, Yang X, Yang H, Dai R, Zeng Q, Jin D. Interaction Analysis of lncRNA and mRNA Based on the Full-Length Transcriptome of the Nymph-to-Adult Developmental Transition of Sogatella furcifera. Insects. 2023; 14(4):308. https://doi.org/10.3390/insects14040308
Chicago/Turabian StyleJia, Zeyan, Xibin Yang, Hong Yang, Renhuai Dai, Qinghui Zeng, and Daochao Jin. 2023. "Interaction Analysis of lncRNA and mRNA Based on the Full-Length Transcriptome of the Nymph-to-Adult Developmental Transition of Sogatella furcifera" Insects 14, no. 4: 308. https://doi.org/10.3390/insects14040308