
Comparative Genomics Reveals Three Genetic Groups of the Whitefly Obligate Endosymbiont Candidatus Portiera aleyrodidarum
 , , and
, , and
Abstract
:Simple Summary
Abstract
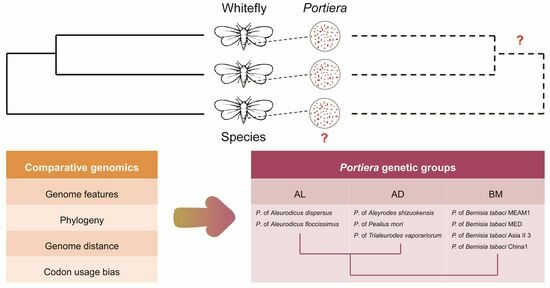
1. Introduction
2. Materials and Methods
2.1. Insect Samples and Genome Sequencing
2.2. Genome Assembly and Annotation
2.3. Phylogenetic Relationships
2.4. Genetic Distance
2.5. Codon Usage Bias
3. Results
3.1. The Genomic Features of Portiera
3.2. The Phylogeny of Portiera and Hosts
3.3. Genetic Distance
3.4. Codon Usage Bias
3.4.1. Synonymous and Nonsynonymous Substitution Rates
3.4.2. PR2 Bias Analysis
3.4.3. Neutrality Plot Analysis
3.4.4. ENC Plot Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Moran, N.A.; Telang, A. Bacteriocyte-associated symbionts of insects. BioScience 1998, 48, 295–304. [Google Scholar] [CrossRef]
- Douglas, A.E. How multi-partner endosymbiosis function. Nat. Rev. Microbiol. 2016, 14, 731–743. [Google Scholar] [CrossRef]
- Bright, M.; Bulgheresi, S. A complex journey: Transmission of microbial symbionts. Nat. Rev. Microbiol. 2010, 8, 218–230. [Google Scholar] [CrossRef]
- Luan, J.-B.; Shan, H.-W.; Isermann, P.; Huang, J.-H.; Lammerding, J.; Liu, S.-S.; Douglas, A.E. Cellular and molecular remodelling of a host cell for vertical transmission of bacterial symbionts. Proc. R. Soc. B 2016, 283, 20160580. [Google Scholar] [CrossRef]
- Moran, N.A.; Baumann, P. Bacterial endosymbionts in animals. Curr. Opin. Microbiol. 2000, 3, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Moeller, A.H.; Sanders, J.G.; Sprockett, D.D.; Landers, A. Assessing co-diversification in host-associated microbiomes. J. Evol. Biol. 2023, online. [Google Scholar] [CrossRef] [PubMed]
- Wernegreen, J.J. Genome evolution in bacterial endosymbionts of insects. Nat. Rev. Genet. 2002, 3, 850–861. [Google Scholar] [CrossRef]
- Ding, H.; Gao, J.; Yang, J.; Zhang, S.; Han, S.; Yi, R.; Ye, Y.; Kan, X. Genome evolution of Buchnera aphidicola (Gammaproteobacteria): Insights into strand compositional asymmetry, codon usage bias, and phylogenetic implications. Int. J. Biol. Macromol. 2023, 253, 126738. [Google Scholar] [CrossRef] [PubMed]
- Santos-Garcia, D.; Latorre, A.; Moya, A.; Gibbs, G.; Hartung, V.; Dettner, K.; Kuechler, S.M.; Silva, F.J. Small but powerful, the primary endosymbiont of moss bugs, Candidatus Evansia muelleri, holds a reduced genome with large biosynthetic capabilities. Genome Biol. Evol. 2014, 6, 1875–1893. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lu, J.; Beattie, G.A.; Islam, M.R.; Om, N.; Dao, H.T.; Nguyen, L.V.; Zaka, S.M.; Guo, J.; Tian, M.; et al. Phylogeography of Diaphorina citri (Hemiptera: Liviidae) and its primary endosymbiont, ‘Candidatus Carsonella ruddii’: An evolutionary approach to host-endosymbiont interaction. Pest Manag. Sci. 2018, 74, 2185–2194. [Google Scholar] [CrossRef]
- Liu, L.; Li, X.-Y.; Huang, X.-L.; Qiao, G.-X. Evolutionary relationships of Pemphigus and allied genera (Hemiptera: Aphididae: Eriosomatinae) and their primary endosymbiont, Buchnera aphidicola. Insect Sci. 2014, 21, 301–312. [Google Scholar] [CrossRef]
- Sloan, D.B.; Moran, N.A. The evolution of genomic instability in the obligate endosymbionts of whiteflies. Genome Biol. Evol. 2013, 5, 783–793. [Google Scholar] [CrossRef]
- Bennett, G.M.; McCutcheon, J.P.; MacDonald, B.R.; Romanovicz, D.; Moran, N.A. Differential genome evolution between companion symbionts in an insect-bacterial symbiosis. mBio 2014, 5, e01697-01614. [Google Scholar] [CrossRef]
- Santos-Garcia, D.; Vargas-Chavez, C.; Moya, A.; Latorre, A.; Silva, F.J. Genome evolution in the primary endosymbiont of whiteflies sheds light on their divergence. Genome Biol. Evol. 2015, 7, 873–888. [Google Scholar] [CrossRef] [PubMed]
- Parvathy, S.T.; Udayasuriyan, V.; Bhadana, V. Codon usage bias. Mol. Biol. Rep. 2022, 49, 539–565. [Google Scholar] [CrossRef]
- Santos-Garcia, D.; Mestre-Rincon, N.; Ouvrard, D.; Zchori-Fein, E.; Morin, S. Portiera gets wild: Genome instability provides insights into the evolution of both whiteflies and their endosymbionts. Genome Biol. Evol. 2020, 12, 2107–2124. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.-R.; Li, N.-N.; Bao, X.-Y.; Douglas, A.E.; Luan, J.-B. Patterns of host cell inheritance in the bacterial symbiosis of whiteflies. Insect Sci. 2020, 27, 938–946. [Google Scholar] [CrossRef]
- Santos-Garcia, D.; Farnier, P.-A.; Beitia, F.; Zchori-Fein, E.; Vavre, F.; Mouton, L.; Moya, A.; Latorre, A.; Silva, F.J. Complete genome sequence of “Candidatus Portiera aleyrodidarum” BT-QVLC, an obligate symbiont that supplies amino acids and carotenoids to Bemisia tabaci. J. Bacteriol. 2012, 194, 6654–6655. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B.; Moran, N.A. Endosymbiotic bacteria as a source of carotenoids in whiteflies. Biol. Lett. 2012, 88, 986–989. [Google Scholar] [CrossRef]
- Zhu, D.-T.; Zou, C.; Ban, F.-X.; Wang, H.-L.; Wang, X.-W.; Liu, Y.-Q. Conservation of transcriptional elements in the obligate symbiont of the whitefly Bemisia tabaci. PeerJ 2019, 7, e7477. [Google Scholar] [CrossRef]
- Chen, C.H.; Dubey, A.K.; Ko, C.C. Comparative morphological studies on two species of Aleyrodes (Hemiptera: Aleyrodidae). Pan-Pac. Entomol. 2007, 83, 244–254. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.; Frati, F.; Beckenbach, A.; Crespi, B.; Liu, H.; Flook, P. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 1994, 87, 651–701. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Tang, H.; Bowers, J.E.; Wang, X.; Ming, R.; Alam, M.; Paterson, A.H. Synteny and collinearity in plant genomes. Science 2008, 320, 486–488. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Chernomor, O.; von Haeseler, A.; Minh, B.Q. Terrace aware data structure for phylogenomic inference from supermatrices. Syst. Biol. 2016, 65, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schremph, D.; Woodhams, M.; von Haeseler, A.; Lanfera, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Hoang, D.H.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2017, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Wu, F.; Cen, Y.; Deng, X.; Zheng, Z.; Chen, J.; Liang, G. The complete mitochondrial genome sequence of Diaphorina citri (Hemiptera: Psyllidae). Mitochondrial DNA B Rseour. 2016, 1, 239–240. [Google Scholar] [CrossRef]
- Wang, H.-L.; Yang, J.; Boykin, L.M.; Zhao, Q.-Y.; Li, Q.; Wang, X.-W.; Liu, S.-S. The characteristics and expression profiles of the mitochondrial genome for the Mediterranean species of the Bemisia tabaci complex. BMC Genom. 2013, 14, 401. [Google Scholar] [CrossRef]
- Chen, S.-C.; Wang, X.-Q.; Li, P.-W.; Hu, X.; Wang, J.-J.; Peng, P. The complete mitochondrial genome of Aleurocanthus camelliae: Insights into gene arrangement and genome organization within the family Aleyrodidae. Int. J. Mol. Sci. 2016, 17, 1843. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-T.; Mu, L.-X.; Wang, J.-R.; Du, Y.-Z. Complete mitochondrial genome of the citrus spiny whitefly Aleurocanthus spiniferus (Quaintance) (Hemiptera: Aleyrodidae): Implications for the phylogeny of whiteflies. PLoS ONE 2016, 11, e0161385. [Google Scholar] [CrossRef]
- Lei, T.; Zhong, Y.-W.; Liu, Y.-Q. Complete mitochondrial genome of the whitefly Aleyrodes shizuokensis Kuwana (Hemiptera: Aleyrodidae), new record from Chinese mainland. Mitochondrial DNA Part B Resour. 2021, 6, 397–398. [Google Scholar] [CrossRef]
- Thao, M.L.; Baumann, P. Evolutionary relationships of primary prokaryotic endosymbionts of whiteflies and their hosts. Appl. Environ. Microb. 2004, 70, 3401–3406. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.-X.; Chen, Z.-T.; Yu, W.-W.; Du, Y.-Z. The complete mitochondrial genome of a spiraling whitefly, Aleurodicus dispersus Russell (Hemiptera: Aleyrodidae). Mitochondrial DNA A DNA Mapp. Seq. Anal. 2017, 28, 165–166. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez, R.L.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Kim, D.; Park, S.; Chun, J. Introducing EzAAI: A pipeline for high throughput calculations of prokaryotic average amino acid identity. J. Microbiol. 2021, 59, 476–480. [Google Scholar] [CrossRef]
- Katz, L.S.; Griswold, T.; Morrison, S.S.; Caravas, J.A.; Zhang, S.; den Bakker, H.C.; Deng, X.; Carleton, H.A. Mashtree: A rapid comparison of whole genome sequence files. J. Open Source Softw. 2019, 4, 1762. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating Gamma-series methods and sliding window strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef]
- Sueoka, N. Intrastrand parity rules of DNA base composition and usage biases of synonymous codons. J. Mol. Evol. 1995, 40, 318–325. [Google Scholar] [CrossRef]
- Sueoka, N. Directional mutation pressure and neutral molecular evolution. Proc. Natl. Acad. Sci. USA 1988, 85, 2653–2657. [Google Scholar] [CrossRef]
- Wright, F. The ‘effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Konstantinidis, K.T.; Tiedje, J.M. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 2567–2572. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.C.; Chimetto, L.; Edwards, R.A.; Swings, J.; Stackebrandt, E.; Thompson, F.L. Microbial genomic taxonomy. BMC Genom. 2013, 14, 913. [Google Scholar] [CrossRef]
- Luan, J.; Sun, X.; Fei, Z.; Douglas, A.E. Maternal inheritance of a single somatic animal cell displayed by the bacteriocyte in the whitefly Bemisia tabaci. Curr. Biol. 2018, 28, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Boyle, L.; O’Neill, S.L.; Robertson, H.M.; Karr, T.L. Interspecific and intraspecific horizontal transfer of Wolbachia in Drosophila. Science 1993, 260, 1796–1799. [Google Scholar] [CrossRef] [PubMed]
- Huigens, M.E.; de Almeida, R.P.; Boons, P.A.; Luck, R.F.; Stouthamer, R. Natural interspecific and intraspecific horizontal transfer of parthenogenesis-inducing Wolbachia in Trichogramma wasps. Proc. Biol. Sci. 2004, 271, 509–515. [Google Scholar] [CrossRef]
- Chong, R.A.; Moran, N.A. Intraspecific genetic variation in hosts affects regulation of obligate heritable symbionts. Proc. Natl. Acad. Sci. USA 2016, 113, 13114–13119. [Google Scholar] [CrossRef]
- Chong, R.A.; Park, H.; Moran, N.A. Genome evolution of the obligate endosymbiont Buchnera aphidicola. Mol. Biol. Evol. 2019, 36, 1481–1489. [Google Scholar] [CrossRef]
- Shigenobu, S.; Watanabe, H.; Hattori, M.; Sakaki, Y.; Ishikawa, H. Genome sequence of the endocellular bacterial symbiont of aphids Buchnera sp. APS. Nature 2000, 407, 81–86. [Google Scholar] [CrossRef]
- Gil, R.; Silva, F.J.; Zientz, E.; Delmotte, F.; Gonzalez-Candelas, F.; Latorre, A.; Rausell, C.; Kamerbeek, J.; Gadau, J.; Holldobler, B.; et al. The genome sequence of Blochmannia floridanus: Comparative analysis of reduced genomes. Proc. Natl. Acad. Sci. USA 2003, 100, 9388–9393. [Google Scholar] [CrossRef] [PubMed]
- Rispe, C.; Delmotte, F.; van Ham, R.C.H.J.; Moya, A. Mutational and selective pressures on codon and amino acid usage in Buchnera, endosymbiotic bacteria of aphids. Genome Res. 2004, 14, 44–53. [Google Scholar] [CrossRef] [PubMed]
- CNCB-NGDC members and partners. Database resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Res. 2022, 50, D27–D38. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Portiera ID | Host Subfamily | Host Species | Accession Number 1 | Reference |
|---|---|---|---|---|
| BTB | Aleyrodinae | Bemisia tabaci MEAM1 | CP003708 | [19] |
| BTQ | Aleyrodinae | Bemisia tabaci MED | CP003835 | [18] |
| BTZ1 | Aleyrodinae | Bemisia tabaci Asia II 3 | CP016327 | [20] |
| BTZ3 | Aleyrodinae | Bemisia tabaci China1 | CP016343 | [20] |
| AdSh | Aleyrodinae | Aleyrodes shizuokensis | GWHBOVO00000000 2 | This study |
| PeMo | Aleyrodinae | Pealius mori | LR744089 | [16] |
| TrVa | Aleyrodinae | Trialeurodes vaporariorum | CP004358 | [12] |
| AlDi | Aleurodicinae | Aleurodicus dispersus | LN649255 | [14] |
| AlFl | Aleurodicinae | Aleurodicus floccissimus | LN734649 | [14] |
| Subfamily | Species | Accession Number | Reference |
|---|---|---|---|
| Aleyrodinae | Bemisia tabaci MEAM1 | KR559508 | NA 1 |
| Aleyrodinae | Bemisia tabaci MED | JQ906700 | [40] |
| Aleyrodinae | Bemisia tabaci China1 | KR559506 | NA 1 |
| Aleyrodinae | Aleurocanthus camelliae | KU761949 | [41] |
| Aleyrodinae | Aleurocanthus spiniferus | KJ437166 | [42] |
| Aleyrodinae | Aleyrodes shizuokensis | MT880225 | [43] |
| Aleyrodinae | Pealius mori | LR877884 | NA 1 |
| Aleyrodinae | Tetraleurodes acaciae | AY521262 | [44] |
| Aleyrodinae | Trialeurodes vaporariorum | AY521265 | [44] |
| Aleurodicinae | Aleurodicus dispersus | KR063274 | [45] |
| Aleurodicinae | Aleurodicus dugesii | AY521251 | [44] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, T.; Luo, N.; Song, C.; Yu, J.; Zhou, Y.; Qi, X.; Liu, Y. Comparative Genomics Reveals Three Genetic Groups of the Whitefly Obligate Endosymbiont Candidatus Portiera aleyrodidarum. Insects 2023, 14, 888. https://doi.org/10.3390/insects14110888
Lei T, Luo N, Song C, Yu J, Zhou Y, Qi X, Liu Y. Comparative Genomics Reveals Three Genetic Groups of the Whitefly Obligate Endosymbiont Candidatus Portiera aleyrodidarum. Insects. 2023; 14(11):888. https://doi.org/10.3390/insects14110888
Chicago/Turabian StyleLei, Teng, Ning Luo, Chao Song, Junwei Yu, Yuhang Zhou, Xin Qi, and Yinquan Liu. 2023. "Comparative Genomics Reveals Three Genetic Groups of the Whitefly Obligate Endosymbiont Candidatus Portiera aleyrodidarum" Insects 14, no. 11: 888. https://doi.org/10.3390/insects14110888