
Cost-Effective PCR-Based Identification of Tunga penetrans (Siphonaptera) Larvae Extracted from Soil Samples Containing PCR Inhibitor-Rich Material
 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
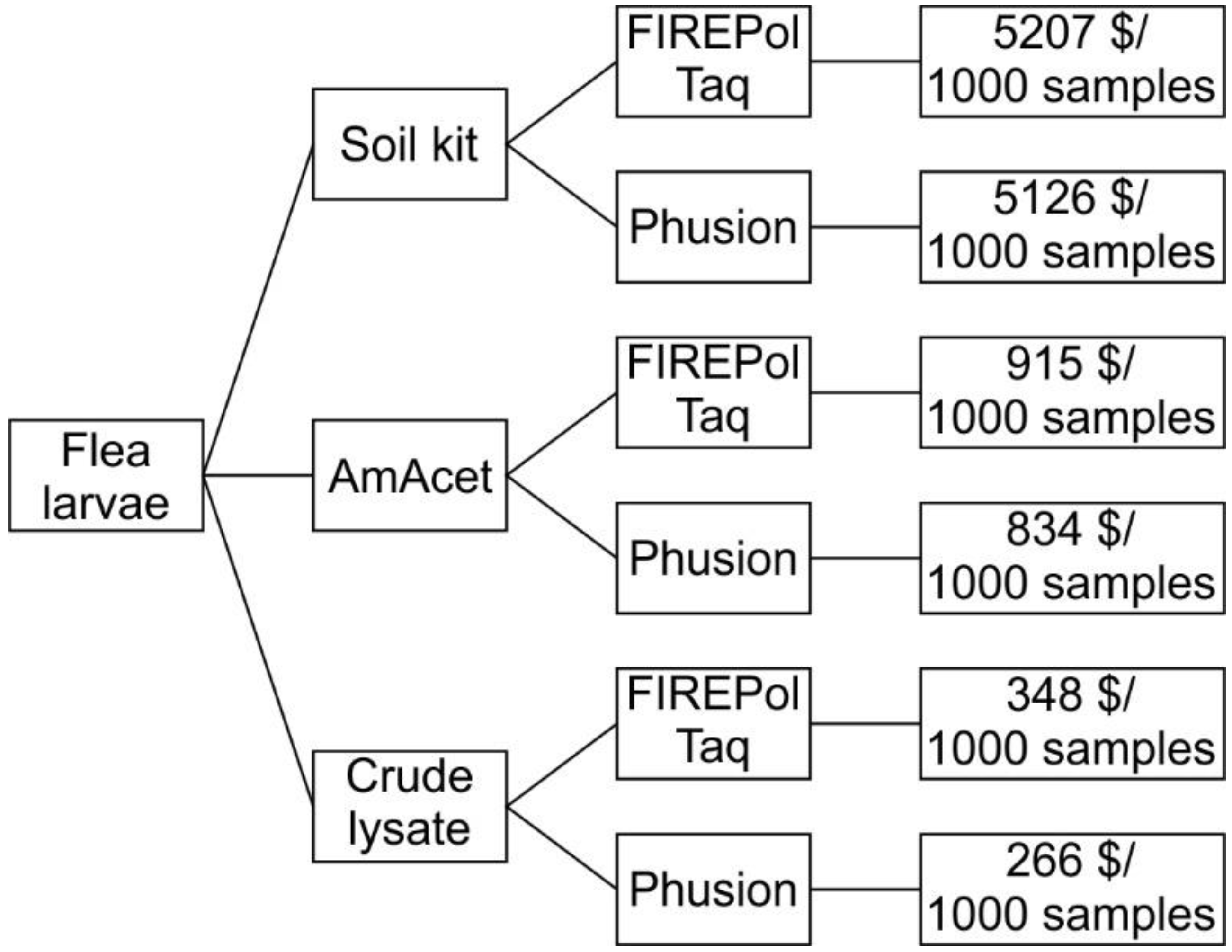
2.1. Experimental Design
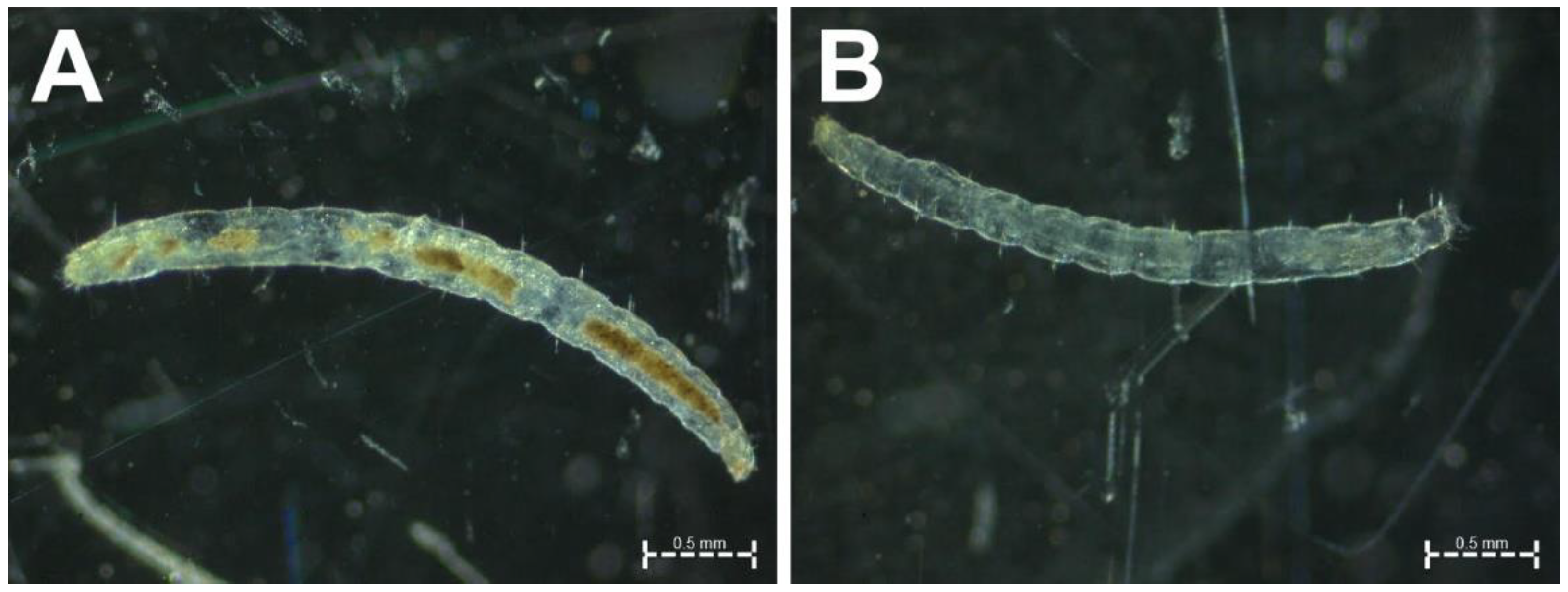
2.2. Flea Sampling
2.3. DNA Preparation Methods
2.3.1. NucleoSpin® Soil DNA Isolation Protocol
2.3.2. Ammonium Acetate DNA Protocol
2.3.3. Crude Flea Lysate Protocol
2.4. PCR Conditions
2.4.1. PCR Primers
2.4.2. GoTaq® DNA Polymerase Conventional PCR Protocol
2.4.3. FIREPol® Taq DNA Polymerase Real-Time PCR Protocol
2.4.4. Phusion® HF DNA Polymerase Real-Time PCR Protocol
2.5. Evaluation of the Specificity of the Tunga penetrans Partial ITS-2 PCR
2.6. PCR Efficacy Analyses for Real-Time PCRs
2.7. Statistical Analyses
3. Results
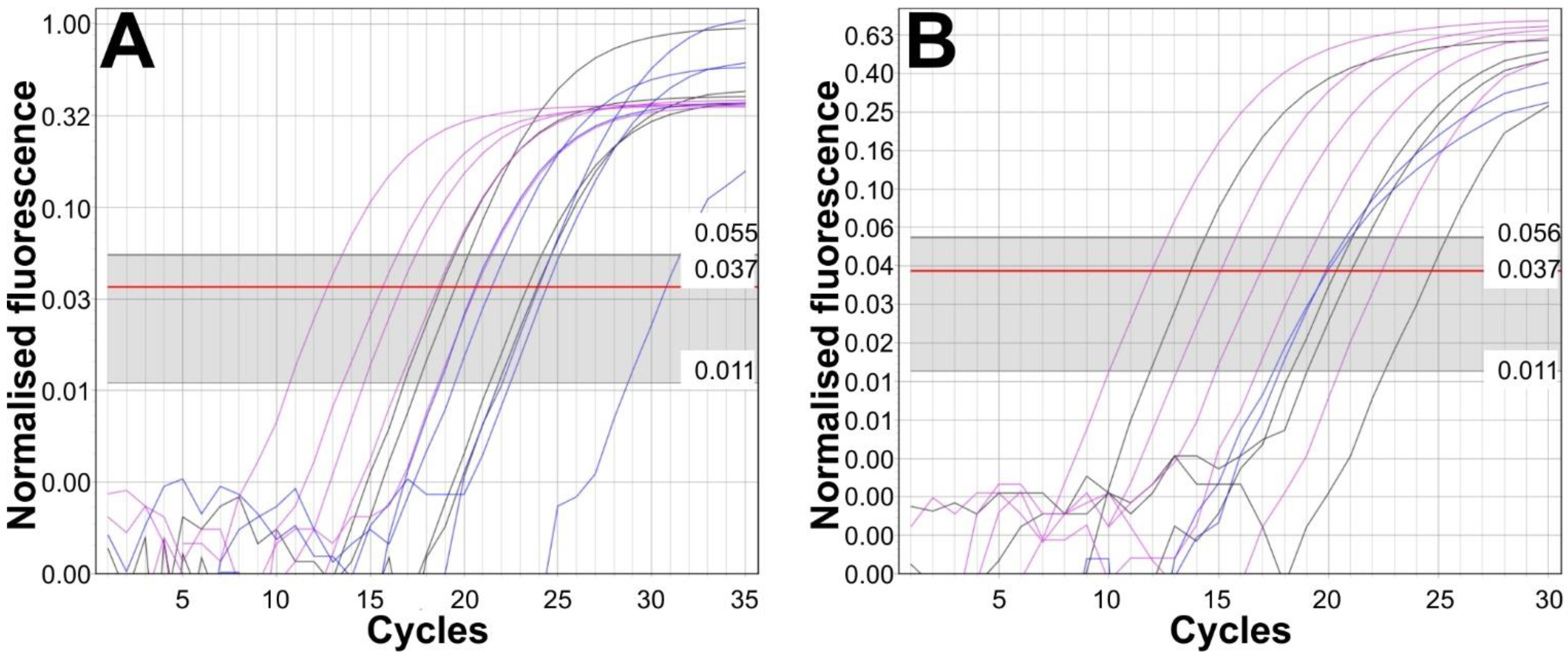
3.1. Initial Comparison of Taq and Phusion®-Based PCR Protocols Using Conventional PCR
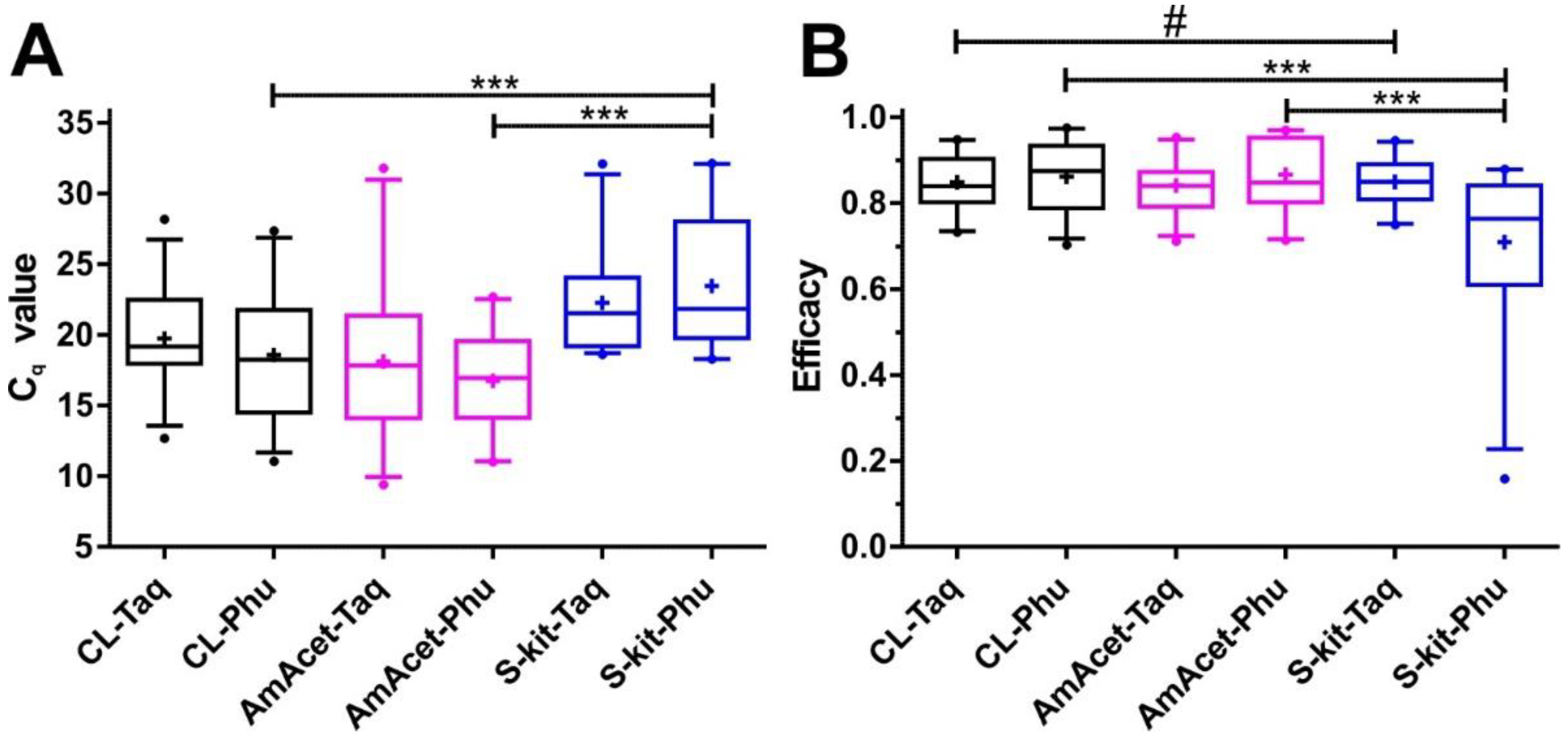
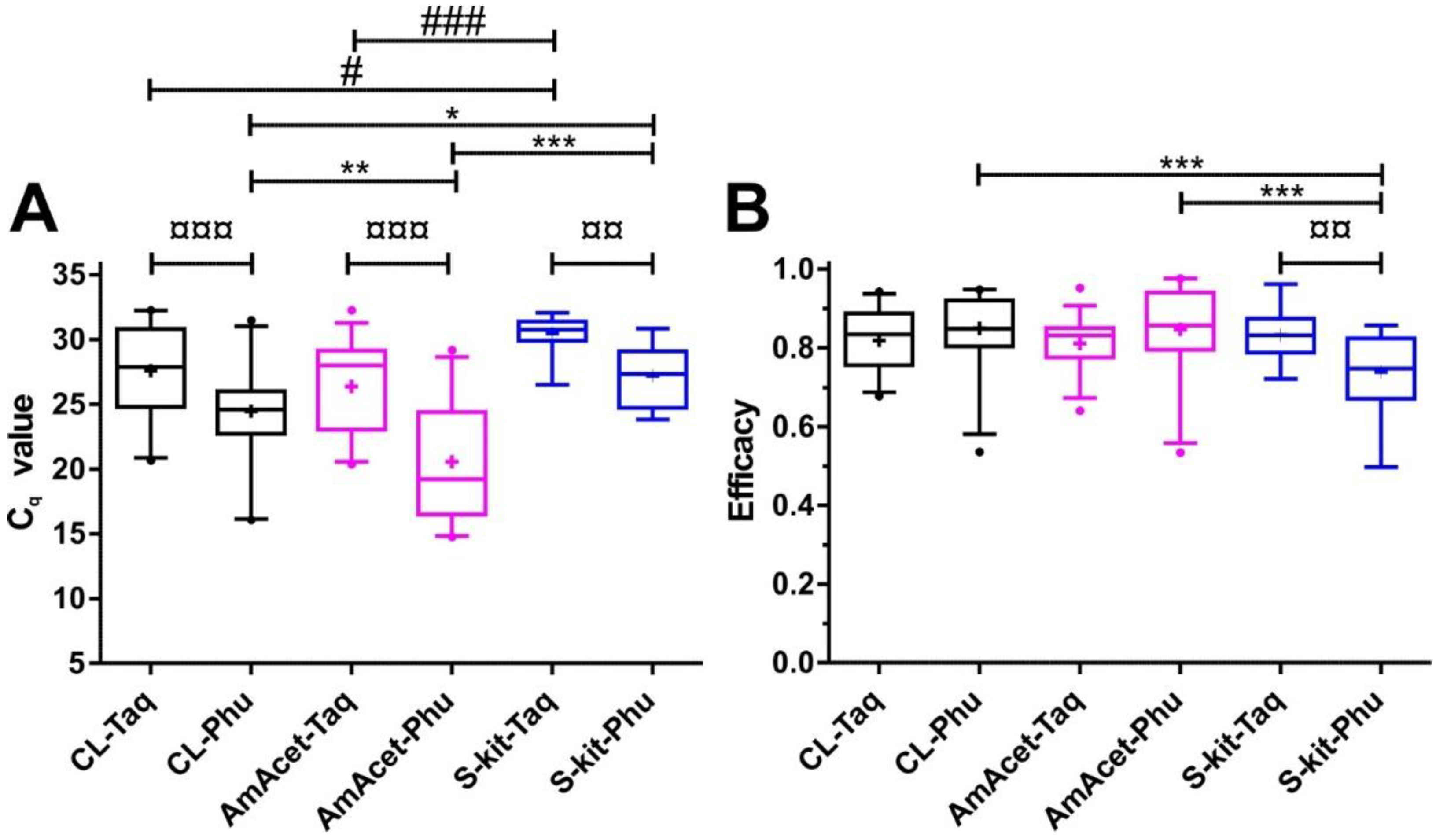
3.2. Comparison of Different Combinations of DNA Preparation and Real-Time PCR Methods
3.2.1. Comparison of Combinations of DNA Preparation Methods and PCR Enzymes Based on a Partial Internal Transcribed Spacer 2 PCR
3.2.2. Comparison of Combinations of DNA Preparation Methods and PCR Enzymes Using cox2-Specific PCR
3.3. Specificity of Tunga penetrans Partial ITS 2 PCR
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feldmeier, H.; Keysers, A. Tungiasis—A Janus-Faced Parasitic Skin Disease. Travel Med. Infect. Dis. 2013, 11, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Mutebi, F.; Krücken, J.; Feldmeier, H.; Von Samson-Himmelstjerna, G. Clinical Implications and Treatment Options of Tungiasis in Domestic Animals. Parasitol. Res. 2021, 120, 4113–4123. [Google Scholar] [CrossRef] [PubMed]
- Harvey, T.V.; Linardi, P.M.; Carlos, R.S.A.; Heukelbach, J. Tungiasis in Domestic, Wild, and Synanthropic Animals in Brazil. Acta Trop. 2021, 222, 106068. [Google Scholar] [CrossRef] [PubMed]
- Feldmeier, H.; Heukelbach, J.; Ugbomoiko, U.S.; Sentongo, E.; Mbabazi, P.; von Samson-Himmelstjerna, G.; Krantz, I. Tungiasis—A Neglected Disease with Many Challenges for Global Public Health. PLoS Negl. Trop. Dis. 2014, 8, e3133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villagrana, S.M.N.; Santisteban, A.G.N. Tungiasis: A Highly Neglected Disease among Neglected Diseases. Case Series from Nduta Refugee Camp (Tanzania). Oxford Med. Case Rep. 2019, 2019, 267–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, H.; Ocampo, J.; Ayala, A.; Trujillo, J.; Feldmeier, H. Very Severe Tungiasis in Amerindians in the Amazon Lowland of Colombia: A Case Series. PLoS Negl. Trop. Dis. 2019, 13, e0007068. [Google Scholar] [CrossRef]
- Mutebi, F.; Krücken, J.; Feldmeier, H.; Waiswa, C.; Mencke, N.; Von Samson-Himmelstjerna, G. Tungiasis-Associated Morbidity in Pigs and Dogs in Endemic Villages of Uganda. Parasites Vectors 2016, 9, 44. [Google Scholar] [CrossRef] [Green Version]
- Mutebi, F.; Krücken, J.; Mencke, N.; Feldmeier, H.; von Samson-Himmelstjerna, G.; Waiswa, C. Two Severe Cases of Tungiasis in Goat Kids in Uganda. J. Insect Sci. 2016, 16, 3–5. [Google Scholar] [CrossRef]
- Mutebi, F.; Krücken, J.; Feldmeier, H.; Waiswa, C.; Mencke, N.; Eneku, W.; von Samson-Himmelstjerna, G. High Intensity of Tunga Penetrans Infection Causing Severe Disease among Pigs in Busoga, South Eastern Uganda. BMC Vet. Res. 2017, 13, 206. [Google Scholar] [CrossRef]
- Harvey, T.V.; dos Santos Freire, Z.; dos Santos, K.C.; de Jesus, A.V.; Guedes, P.E.B.; da Paixão Sevá, A.; de Almeida Borges, F.; Carlos, R.S.A. Clinical and Macroscopic Morphological Features of Canine Tungiasis. Parasitol. Res. 2021, 120, 807–818. [Google Scholar] [CrossRef]
- Bitam, I.; Dittmar, K.; Parola, P.; Whiting, M.F.; Raoult, D. Fleas and Flea-Borne Diseases. Int. J. Infect. Dis. 2010, 14, e667–e676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durden, L.A.; Hinkle, N.C. Fleas (Siphonaptera). In Medical and Veterinary Entomology, 3rd ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 145–169. [Google Scholar] [CrossRef]
- Hsu, M.H.; Hsu, Y.C.; Wu, W.J. Consumption of Flea Faeces and Eggs by Larvae of the Cat Flea, Ctenocephalides Felis. Med. Vet. Entomol. 2002, 16, 445–447. [Google Scholar] [CrossRef] [PubMed]
- Poulin, R.; Krasnov, B.R.; Shenbrot, G.I.; Mouillot, D.; Khokhlova, I.S. Evolution of Host Specificity in Fleas: Is It Directional and Irreversible? Int. J. Parasitol. 2006, 36, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Coates, S.J.; Thomas, C.; Chosidow, O.; Engelman, D.; Chang, A.Y. Ectoparasites. J. Am. Acad. Dermatol. 2020, 82, 551–569. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Xu, S.; Yu, Z.; Li, N.; Wang, R.; Gao, X.; Yang, X.; Liu, J. Abundance and Seasonal Activity of Haemaphysalis Concinna (Acari: Ixodidae) at the Border between China and Russia in Northern Inner Mongolia, China. Parasites Vectors 2016, 9, 1. [Google Scholar] [CrossRef] [Green Version]
- Krasnov, B.R.; Khokhlova, I.S.; Fielden, L.J.; Burdelova, N.V. Development Rates of Two Xenopsylla Flea Species in Relation to Air Temperature and Humidity. Med. Vet. Entomol. 2001, 15, 249–258. [Google Scholar] [CrossRef]
- Krasnov, B.R.; Khokhlova, I.S.; Fielden, L.J.; Burdelova, N.V. Effect of Air Temperature and Humidity on the Survival of Pre-Imaginal Stages of Two Flea Species (Siphonaptera: Pulicidae). J. Med. Entomol. 2001, 38, 629–637. [Google Scholar] [CrossRef]
- Rust, M.K. Recent Advancements in the Control of Cat Fleas. Insects 2020, 11, 668. [Google Scholar] [CrossRef]
- Hoeppli, R. Early References to the Occurrence of Tunga Penetrans in Tropical Africa. Acta Trop. 1963, 20, 143–153. [Google Scholar]
- Deka, M.A. Mapping the Geographic Distribution of Tungiasis in Sub-Saharan Africa. Trop. Med. Infect. Dis. 2020, 5, 122. [Google Scholar] [CrossRef]
- Matheson, C.D.; Gurney, C.; Esau, N.; Lehto, R. Assessing PCR Inhibition from Humic Substances. Open Enzym. Inhib. J. 2010, 3, 38–45. [Google Scholar] [CrossRef] [Green Version]
- Schrader, C.; Schielke, A.; Ellerbroek, L.; Johne, R. PCR Inhibitors—Occurrence, Properties and Removal. J. Appl. Microbiol. 2012, 113, 1014–1026. [Google Scholar] [CrossRef] [PubMed]
- Moreira, D. Efficient Removal of PCR Inhibitors Using Agarose-Embedded DNA Preparations. Nucleic Acids Res. 1998, 26, 3309–3310. [Google Scholar] [CrossRef] [PubMed]
- McInerney, P.; Adams, P.; Hadi, M.Z. Error Rate Comparison during Polymerase Chain Reaction by DNA Polymerase. Mol. Biol. Int. 2014, 2014, 287430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, M.; Tanigawa, C.; Fujii, Y.; Kaneko, S. Comparação de Seis Polimerases de DNA Disponíveis Comercialmente Para o PCR Direto. Rev. Inst. Med. Trop. Sao Paulo 2013, 55, 401–406. [Google Scholar] [CrossRef]
- Trombley Hall, A.; McKay Zovanyi, A.; Christensen, D.R.; Koehler, J.W.; Devins Minogue, T. Evaluation of Inhibitor-Resistant Real-Time PCR Methods for Diagnostics in Clinical and Environmental Samples. PLoS ONE 2013, 8, e73845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demeler, J.; Ramünke, S.; Wolken, S.; Ianiello, D.; Rinaldi, L.; Gahutu, J.B.; Cringoli, G.; von Samson-Himmelstjerna, G.; Krücken, J. Discrimination of Gastrointestinal Nematode Eggs from Crude Fecal Egg Preparations by Inhibitor-Resistant Conventional and Real-Time PCR. PLoS ONE 2013, 8, e61285. [Google Scholar] [CrossRef] [Green Version]
- González, G.; Barberena-Arias, M.F.; Huang, W.; Ospina-Sánchez, C.M. Sampling Methods for Soil and Litter Fauna. In Measuring Arthropod Biodiversity; Santos, J.C., Fernandes, G.W., Eds.; Springer: Cham, Switzerland, 2021. [Google Scholar] [CrossRef]
- Mutebi, F.; Krücken, J.; Feldmeier, H.; Waiswa, C.; Mencke, N.; Sentongo, E.; von Samson-Himmelstjerna, G. Animal Reservoirs of Zoonotic Tungiasis in Endemic Rural Villages of Uganda. PLoS Negl. Trop. Dis. 2015, 9, e0004126. [Google Scholar] [CrossRef] [Green Version]
- Whiting, M.F. Mecoptera Is Paraphyletic: Multiple Genes and Phylogeny of Mecoptera and Siphonaptera. Zool. Scr. 2002, 31, 93–104. [Google Scholar] [CrossRef]
- Hornok, S.; Beck, R.; Farkas, R.; Grima, A.; Otranto, D.; Kontschán, J.; Takács, N.; Horváth, G.; Szoke, K.; Szekeres, S.; et al. High Mitochondrial Sequence Divergence in Synanthropic Flea Species (Insecta: Siphonaptera) from Europe and the Mediterranean. Parasites Vectors 2018, 11, 221. [Google Scholar] [CrossRef] [Green Version]
- Luchetti, A.; Mantovani, B.; Pampiglione, S.; Trentini, M. Molecular Characterization of Tunga Trimamillata and T. Penetrans (Insecta, Siphonaptera, Tungidae): Taxonomy and Genetic Variability. Parasite 2005, 12, 123–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dryden, M.W.; Rust, M.K. The Cat Flea: Biology, Ecology and Control. Vet. Parasitol. 1994, 52, 1–19. [Google Scholar] [CrossRef] [PubMed]
- De Avelar, D.M.; Facury Filho, E.J.; Linardi, P.M. A New Species of Tunga (Siphonaptera: Tungidae) Parasitizing Cattle from Brazil. J. Med. Entomol. 2013, 50, 679–684. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Primer Name | Primer Sequence (5′->3′) | Size (abp) | Annealing Temperatures (°C) | Reference |
|---|---|---|---|---|---|
| Tunga penetrans ITS-2 | TPS-F | TGCTCGACCCGGTGACGGGA | 278 | FIREPol® Taq 65 Phusion® 69 | This study |
| TPS-R | CGCGCAAAGCGTGGAGGTTTCG | ||||
| Cox2 | F-Leu | TCTAATATGGCAGATTAGTGC | 730 | GoTaq 53FIREPol® Taq 53 Phusion® 53 | [32] |
| R-Lys | GAGACCAGTACTTGCTTTCAGTCATC |
| FIREPol® Taq | Phusion® | p Value | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PCR | DNA Preparation | n | % pos. | 95% CI | n | % pos. | 95% CI | FIREPol® vs. Phusion® a | FIREPol b | Phusion® b |
| Tunga penetrans partial ITS-2 | ||||||||||
| S-kit | 28 | 93.3 | 78.7–98.1 | 24 | 80.0 | 62.7–90.5 | 0.153 | 1 | 0.057 | |
| AmAcet. | 28 | 93.3 | 78.7–98.1 | 29 | 96.7 | 83.2–99.4 | 0.619 | 0.433 | 0.753 | |
| CL | 26 | 86.7 | 70.3–94.7 | 25 | 83.3 | 66.4–92.7 | 0.736 | 0.433 | 0.109 | |
| Cox2 | ||||||||||
| S-kit | 17 | 56.7 | 39.1–72.6 | 17 | 56.7 | 39.1–72.6 | 1 | <0.001 | 0.191 | |
| AmAcet. | 30 | 100 | 88.7–100 | 22 | 73.3 | 55.6–85.8 | 0.002 | 0.112 | 0.112 | |
| CL | 23 | 76.7 | 59.1–88.2 | 23 | 76.7 | 59.1–88.2 | 1 | 0.005 | 0.776 | |
| Target Quantity (Copy Numbers) | 1 × 105, a | 1 × 103, a | 1 × 101, a |
|---|---|---|---|
| Cq Value (Mean (range)) | Cq Value (Mean (range)) | Cq Value (Mean (range)) | |
| Target Species | |||
| T. penetrans | 22.49 (2.946) | 25.81 (5.239) | 28.46 (3.198) |
| T. trimamillata | n.a. | n.a. | n.a. |
| C. felis (genomic DNA) b | n.a. | n.a. | n.a. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amugune, B.L.; Matharu, A.K.; Ouma, P.; Mutebi, F.; Elson, L.; Fillinger, U.; Krücken, J. Cost-Effective PCR-Based Identification of Tunga penetrans (Siphonaptera) Larvae Extracted from Soil Samples Containing PCR Inhibitor-Rich Material. Insects 2023, 14, 5. https://doi.org/10.3390/insects14010005
Amugune BL, Matharu AK, Ouma P, Mutebi F, Elson L, Fillinger U, Krücken J. Cost-Effective PCR-Based Identification of Tunga penetrans (Siphonaptera) Larvae Extracted from Soil Samples Containing PCR Inhibitor-Rich Material. Insects. 2023; 14(1):5. https://doi.org/10.3390/insects14010005
Chicago/Turabian StyleAmugune, Billy L., Abneel K. Matharu, Paul Ouma, Francis Mutebi, Lynne Elson, Ulrike Fillinger, and Jürgen Krücken. 2023. "Cost-Effective PCR-Based Identification of Tunga penetrans (Siphonaptera) Larvae Extracted from Soil Samples Containing PCR Inhibitor-Rich Material" Insects 14, no. 1: 5. https://doi.org/10.3390/insects14010005