
Transcriptome Analysis of Beet Webworm Shows That Histone Deacetylase May Affect Diapause by Regulating Juvenile Hormone

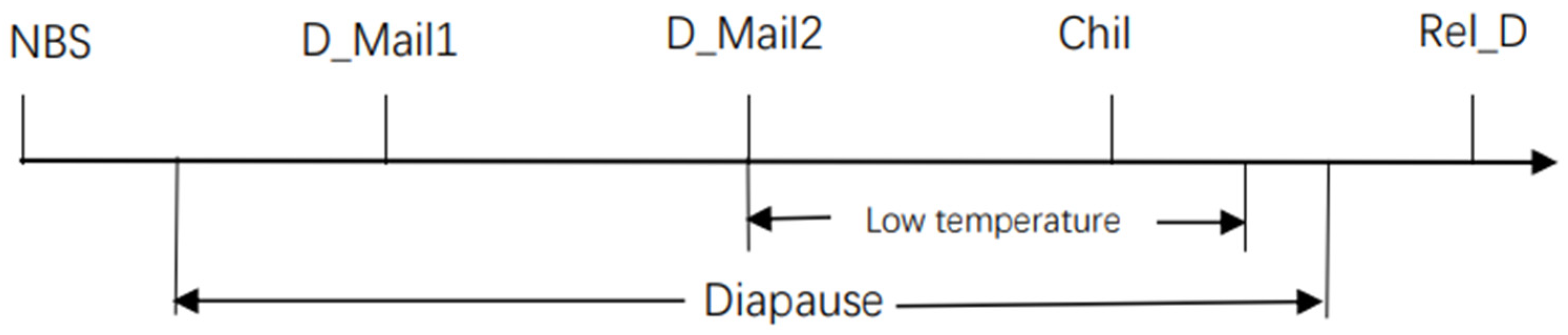{kind=link}
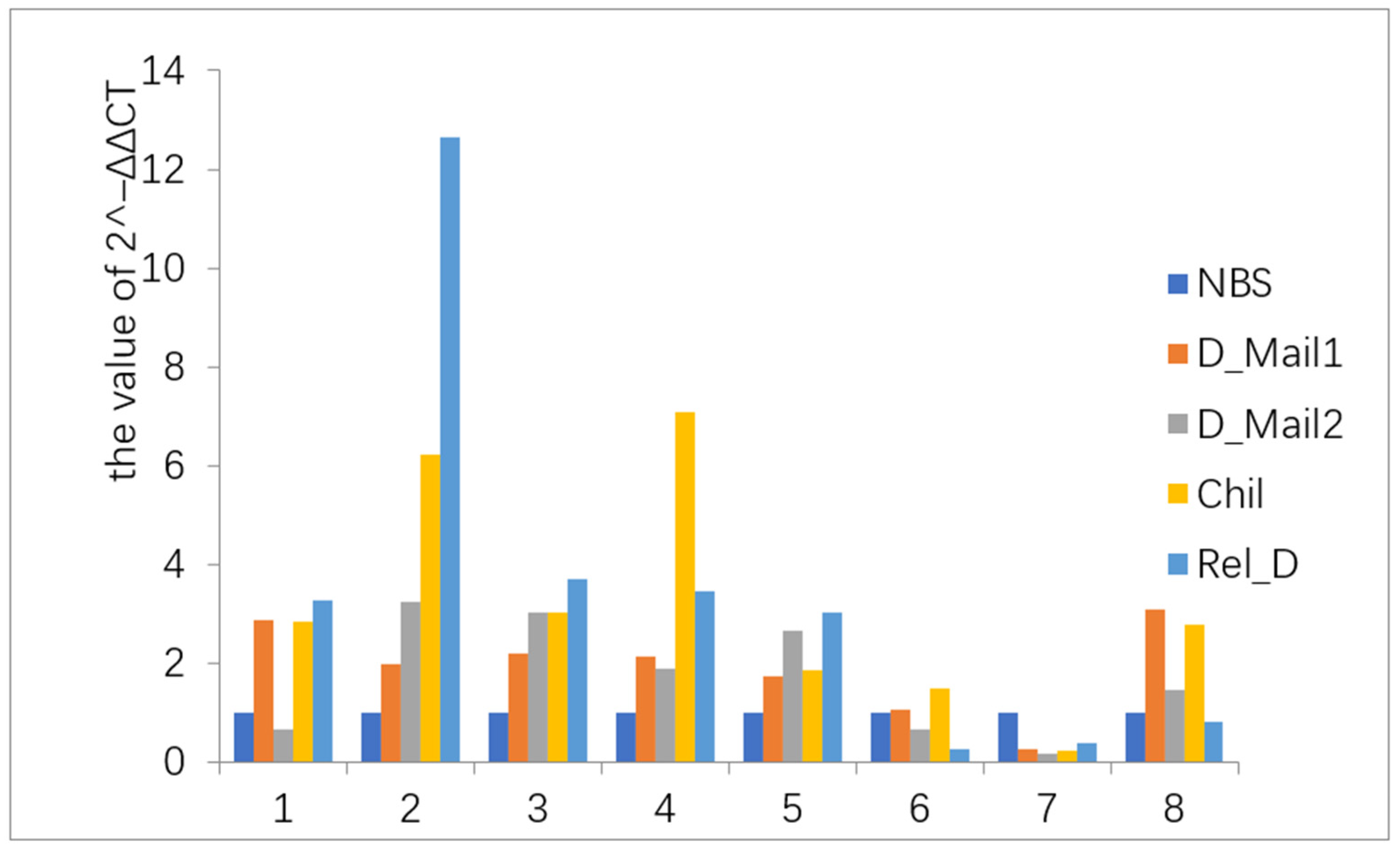{kind=link}
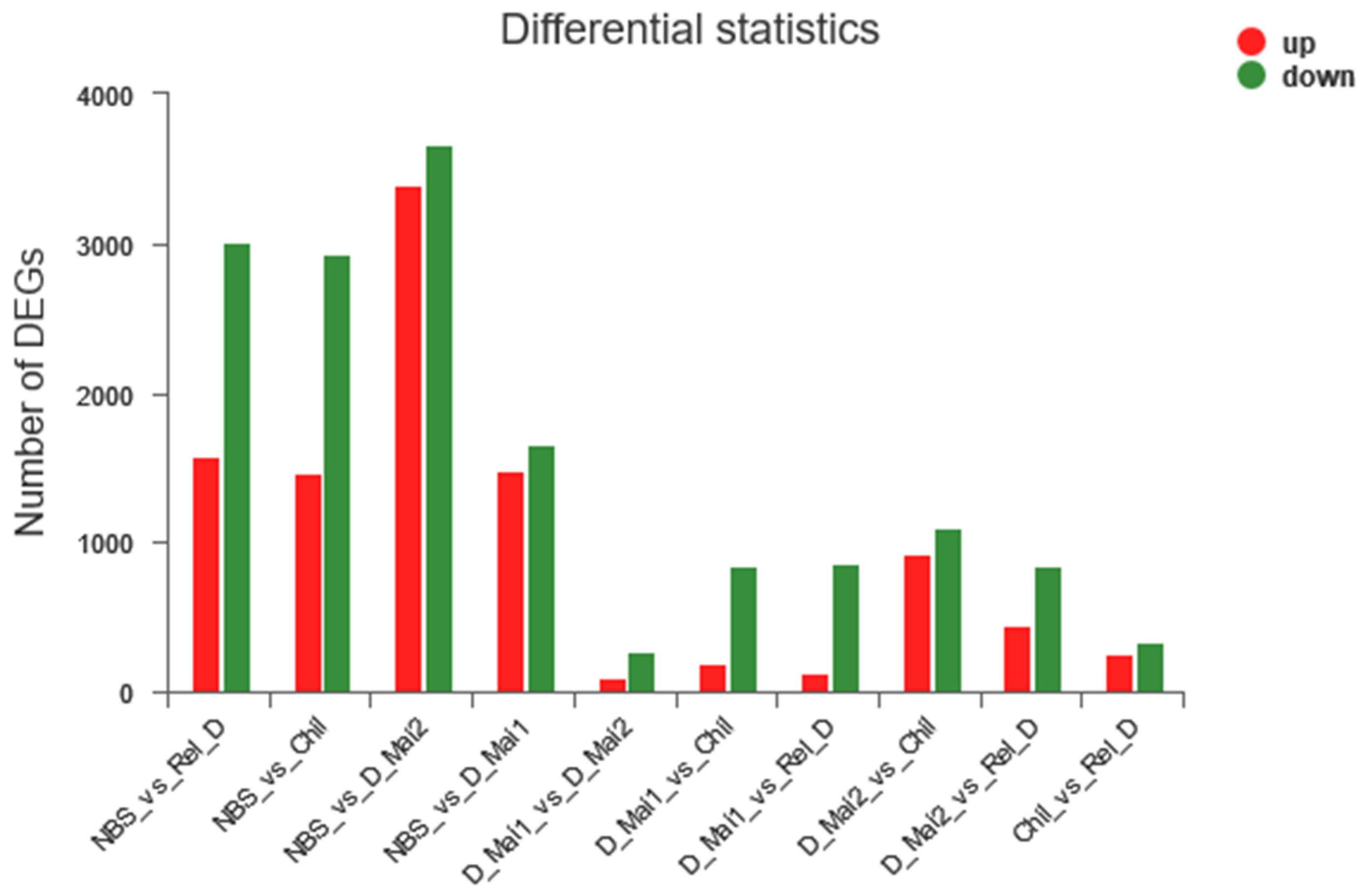{kind=link}
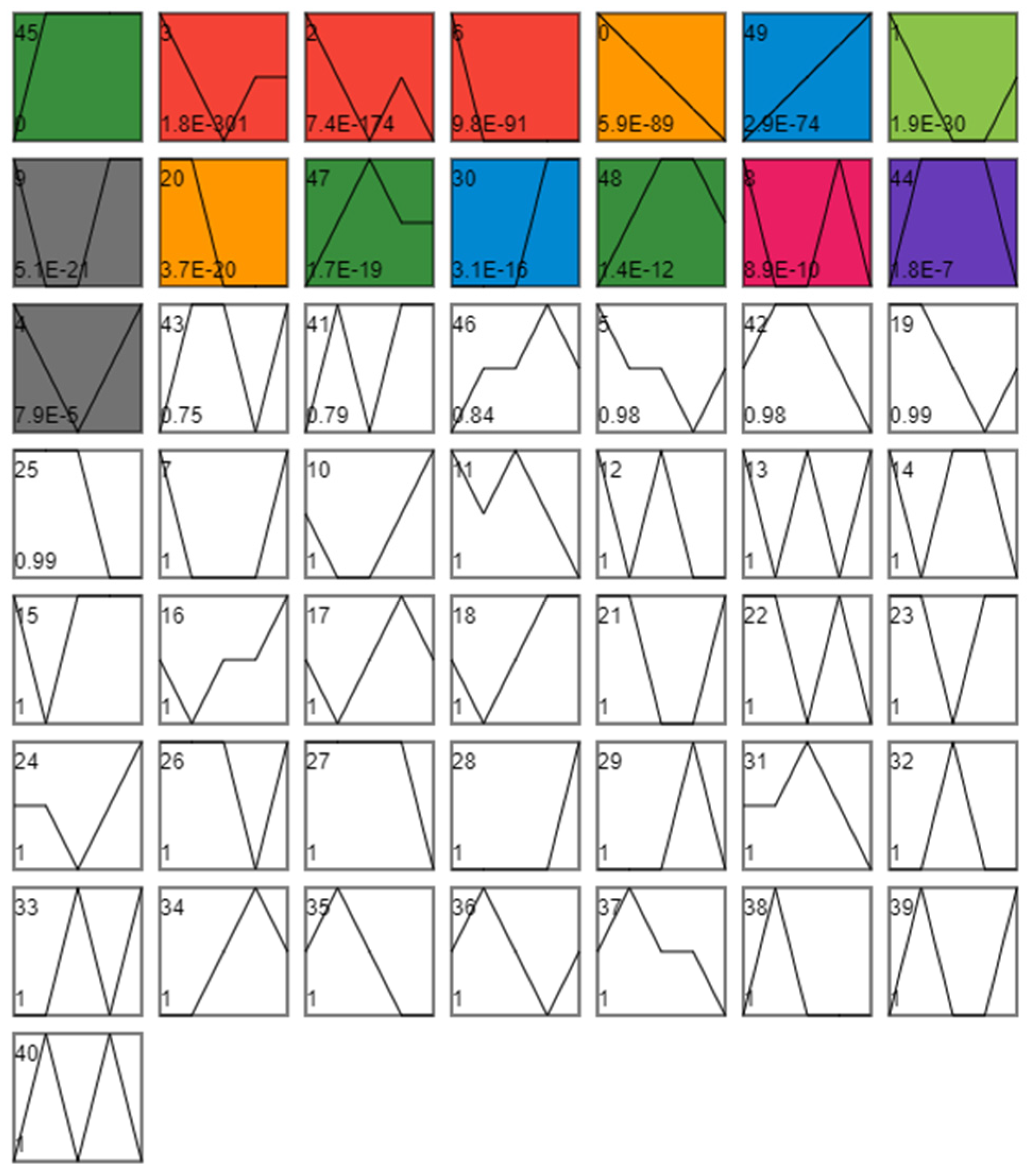{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Insects
2.2. Transcriptome Sequencing and Bioinformatics Analysis
2.2.1. Nucleic Acid Extraction
2.2.2. Library Preparation, Illumina Sequencing, De Novo Assembly and Annotation
2.2.3. Differential Expression Analysis and Functional Enrichment
2.2.4. Time Series Analysis
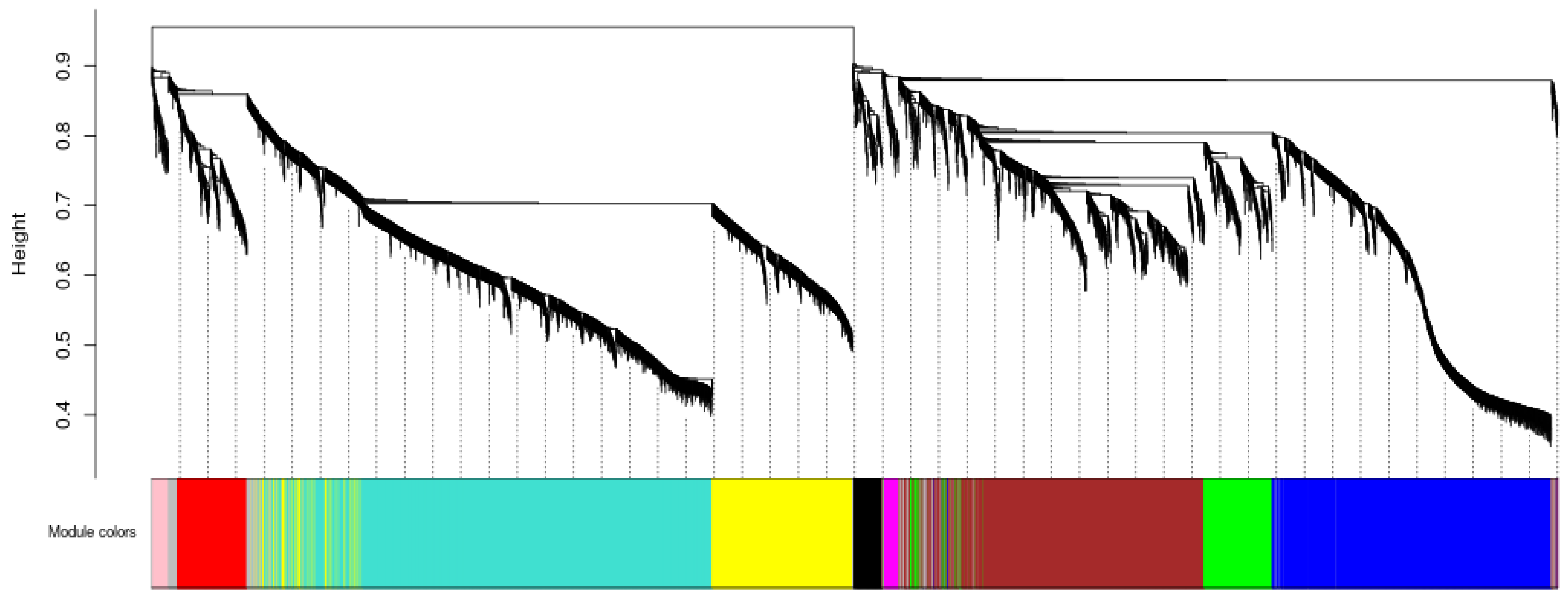
2.2.5. Identification of DEGSs and Co-Expression Network Module
2.2.6. Gene Set Enrichment Analysis
2.3. Quantitative Reverse Transcription RT-PCR Analysis
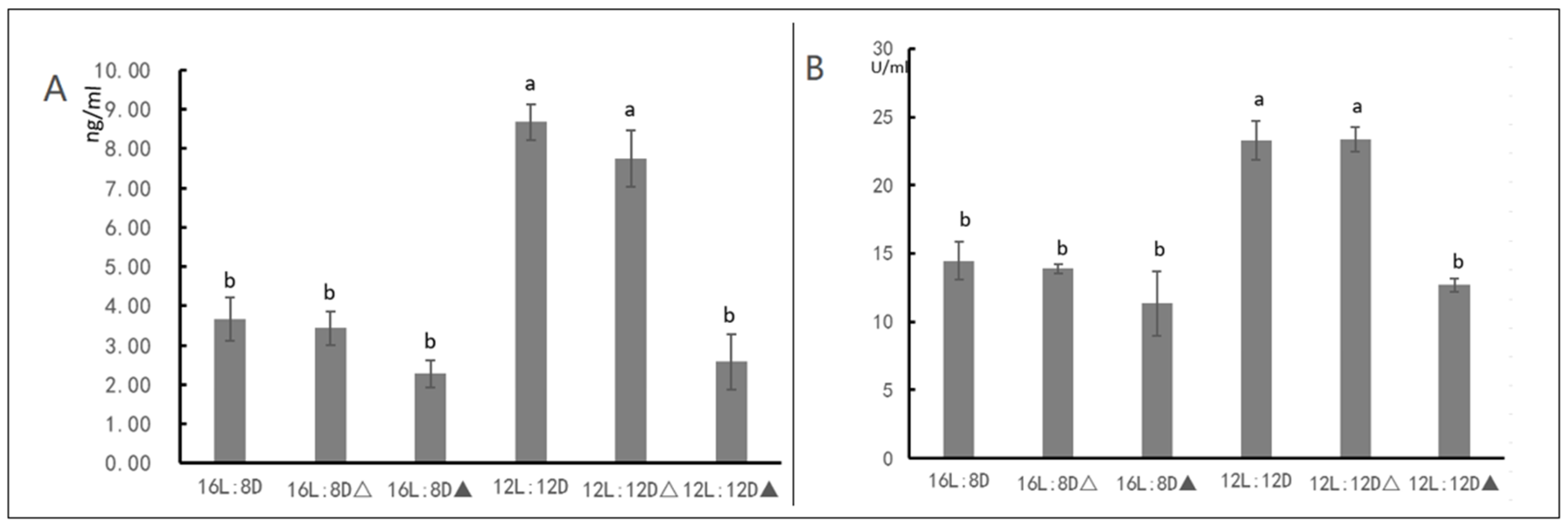
2.4. Measurement of JH content and HDAC Enzyme Activity
2.5. Injection of HDAC Inhibitor into Beet Webworm
3. Results
3.1. Simple Analysis of Transcriptome Data
3.1.1. Identification of DEGs
3.1.2. Analysis of Differences in Time Series Expression
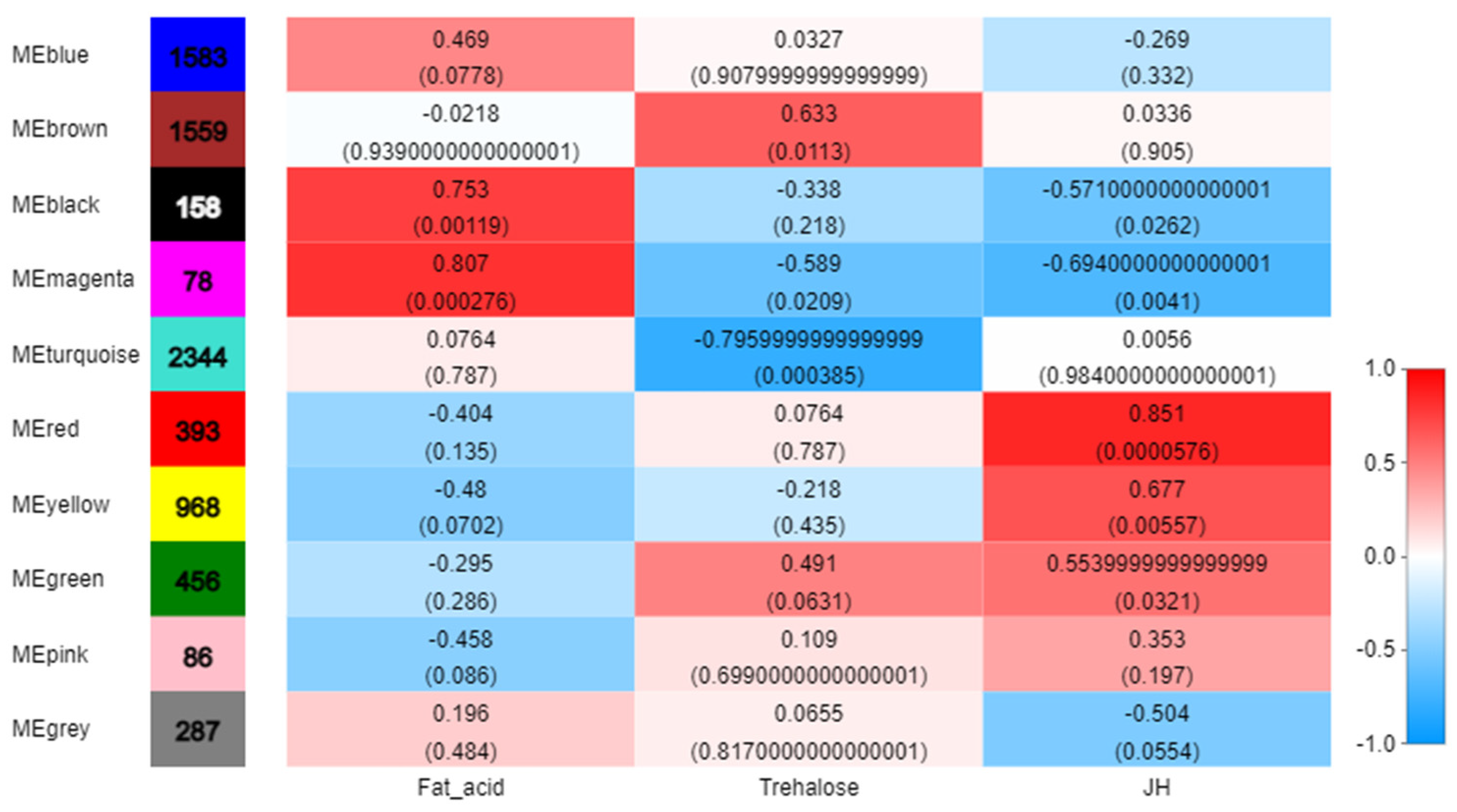
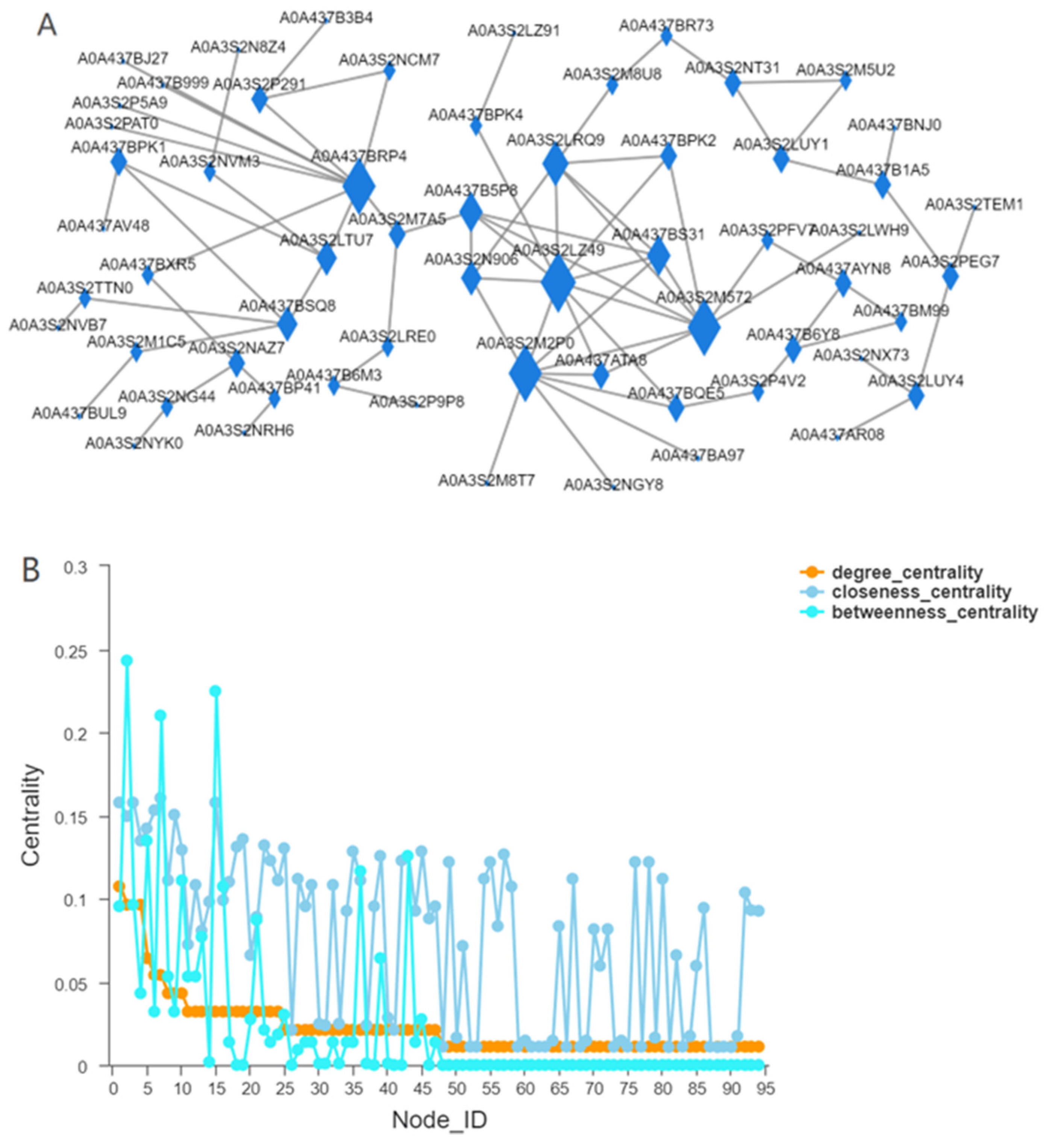
3.2. Identification of WGCNA Modules Associated with JH
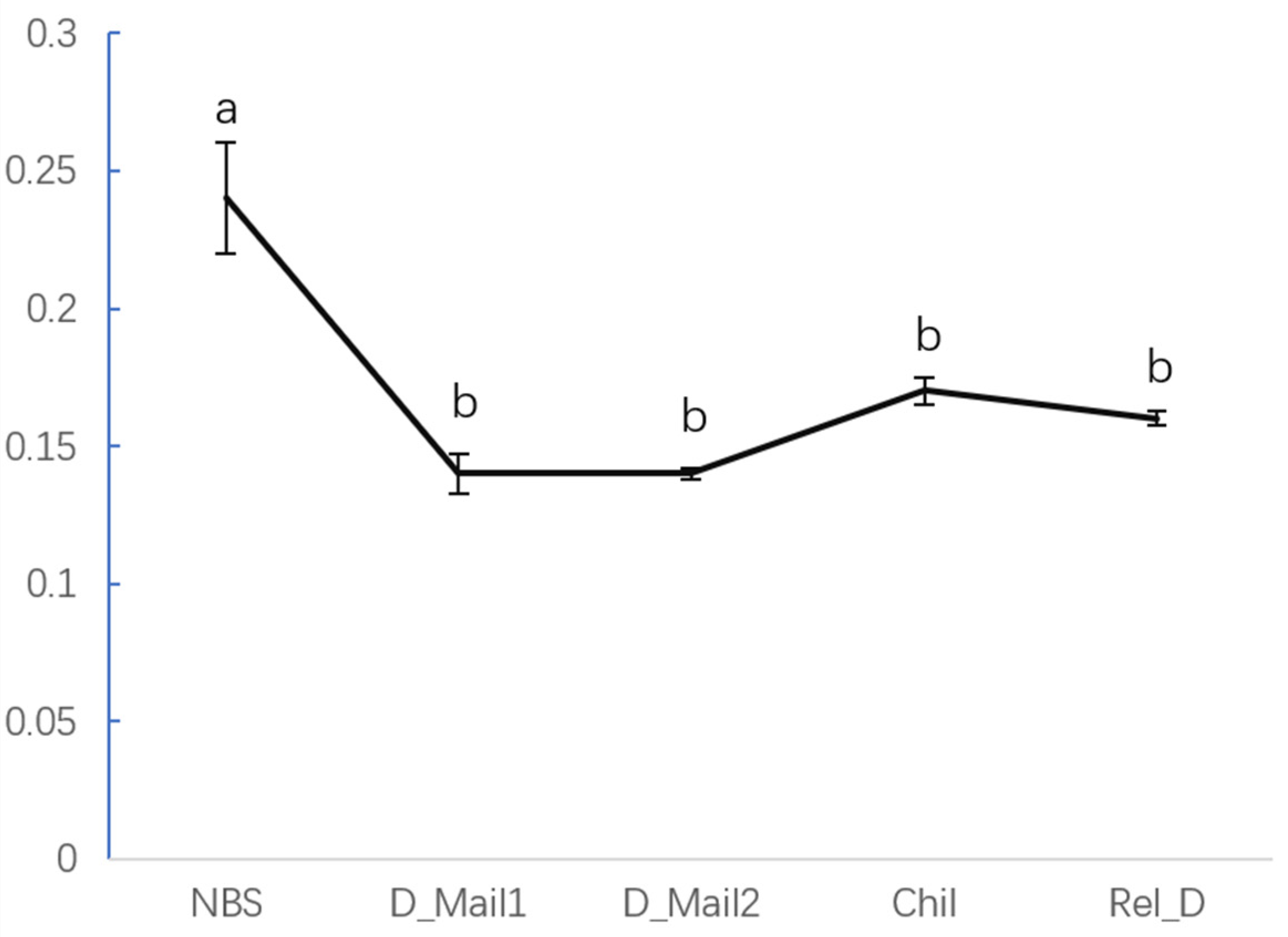
3.3. HDAC Inhibitor Suggests That HDAC Affect JH Content
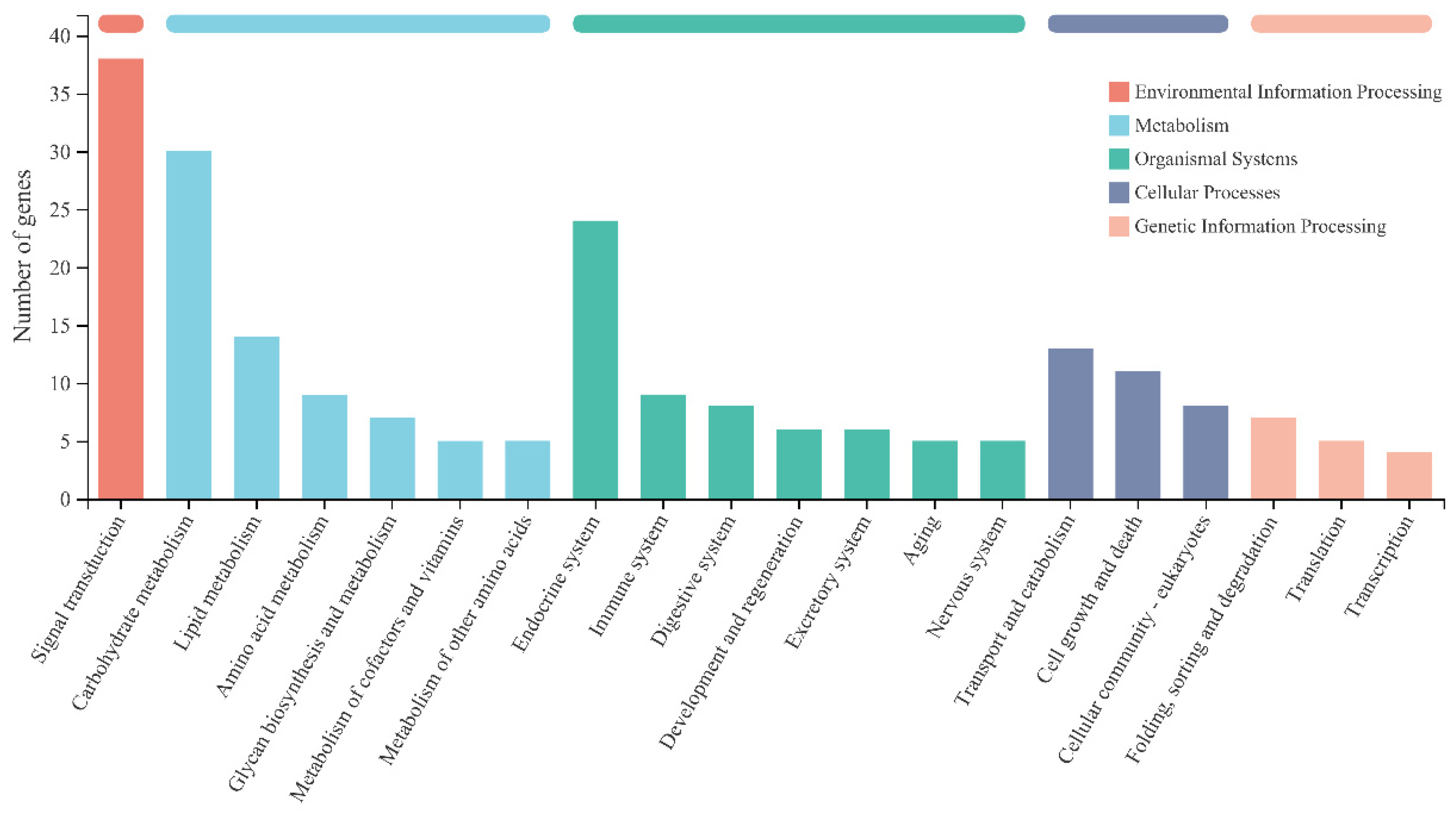
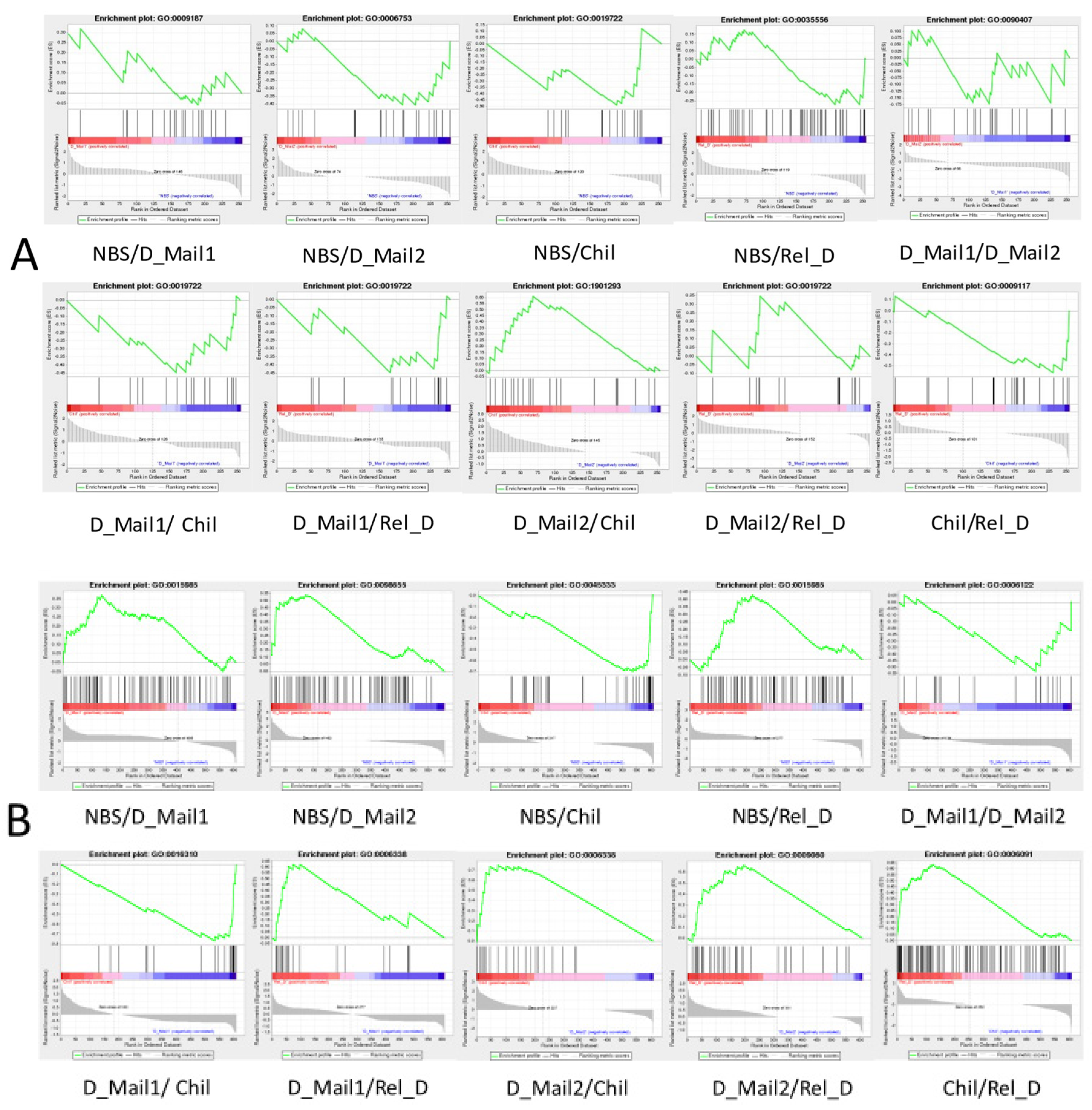
3.4. GSEA Results Showed a Significant Effect of Thermogenic Genes on Diapause
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feng, H.; Wu, K.; Cheng, D.; Guo, Y. Spring migration and summer dispersal of Loxostege sticticalis (Lepidoptera: Pyralidae) and other insects observed with radar in northern China. Environ. Entomol. 2004, 33, 1253–1265. [Google Scholar] [CrossRef]
- Frolov, A.N. The beet webworm Loxostege sticticalis L. (Lepidoptera, Crambidae) in the focus of agricultural entomology objectives: I. The periodicity of pest outbreaks. Entomol. Rev. 2015, 95, 147–156. [Google Scholar] [CrossRef]
- Cheng, Y.; Luo, L.; Sappington, T.W.; Jiang, X.; Zhang, L.; Frolov, A.N. Onset of Oviposition Triggers Abrupt Reduction in Migratory Flight Behavior and Flight Muscle in the Female Beet Webworm, Loxostege sticticalis. PLoS ONE 2016, 11, e0166859. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Huang, S.; Luo, L. Juvenile hormone changes associated with diapause induction, maintenance, and termination in the beet webworm, Loxostege sticticalis (Lepidoptera: Pyralidae). Arch. Insect Biochem. Physiol. 2011, 77, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.; Cheng, Y.; Luo, L.; Sappington, T.W.; Jiang, X.; Zhang, L. Density-dependent prophylaxis in crowded Beet Webworm, Loxostege sticticalis(Lepidoptera: Pyralidae) larvae to a parasitoid and a fungal pathogen. Int. J. Pest Manag. 2013, 59, 174–179. [Google Scholar] [CrossRef]
- Danks, H.V. Insect Dormancy: An Ecological Perspective; Biological Survey of Canada (Terrestrial Arthropods): Ottawa, ON, Canada, 1987. [Google Scholar]
- Xie, D.; Luo, L.; Sappington, T.W.; Jiang, X.; Zhang, L. Comparison of reproductive and flight capacity of Loxostege sticticalis (Lepidoptera: Pyralidae), developing from diapause and non-diapause larvae. Environ. Entomol. 2012, 41, 1199–1207. [Google Scholar] [CrossRef]
- Denlinger, D.L. Regulation of diapause. Annu. Rev. Entomol. 2002, 47, 93. [Google Scholar] [CrossRef]
- Saunders, D.; Richard, D.; Applebaum, S.; Ma, M.; Gilbert, L.J.G. Photoperiodic diapause in Drosophila melanogaster involves a block to the juvenile hormone regulation of ovarian maturation. Gen. Comp. Endocrinol. 1990, 79, 174–184. [Google Scholar] [CrossRef]
- Yamashita, O. Diapause hormone of the silkworm, Bombyx mori: Structure, gene expression and function. J. Insect Physiol. 1996, 42, 669–679. [Google Scholar] [CrossRef]
- Cho, J.R.; Lee, M.; Kim, H.S.; Boo, K.S. Effect of the juvenile hormone analog, fenoxycarb on termination of reproductive diapause in Scotinophara lurida (Burmeister) (Heteroptera: Pentatomidae). J. Asia Pac. Entomol. 2007, 10, 145–150. [Google Scholar] [CrossRef]
- Liu, W.; Li, Y.; Zhu, L.; Zhu, F.; Lei, C.-L.; Wang, X.-P. Juvenile hormone facilitates the antagonism between adult reproduction and diapause through the methoprene-tolerant gene in the female Colaphellus bowringi. Insect Biochem. Mol. Biol. 2016, 74, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Denlinger, D.L.; Yocum, G.D.; Rinehart, J.P. 10—Hormonal Control of Diapause. In Insect Endocrinology; Academic Press: Cambridge, MA, USA, 2012; pp. 430–463. [Google Scholar]
- Henry, H.L.; Norman, A.W. (Eds.) Encyclopedia of Hormones; Elsevier Science: Amsterdam, The Netherlands, 2003; pp. 528–535. [Google Scholar] [CrossRef]
- Bogus, M.; Scheller, K. Allatotropin released by the brain controls larval molting in Galleria mellonella by affecting juvenile hormone synthesis. Int. J. Dev. Biol. 1996, 40, 205–210. [Google Scholar] [PubMed]
- Hartfelder, K.; Emlen, D.J. Endocrine control of insect polyphenism. In Insect Endocrinology; Academic Press: Cambridge, MA, USA, 2012; pp. 464–522. [Google Scholar] [CrossRef]
- Glastad, K.M.; Hunt, B.G.; Goodisman, M.A.D. Epigenetics in Insects: Genome Regulation and the Generation of Phenotypic Diversity. Annu. Rev. Entomol. 2019, 64, 185–203. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.A.; Bautista-Jimenez, R.; Denlinger, D.L. Changes in histone acetylation as potential mediators of pupal diapause in the flesh fly, Sarcophaga bullata. Insect Biochem. Mol. Biol. 2016, 76, 29–37. [Google Scholar] [CrossRef]
- Reynolds, J.A.; Hand, S.C. Embryonic diapause highlighted by differential expression of mRNAs for ecdysteroidogenesis, transcription and lipid sparing in the cricket Allonemobius socius. J. Exp. Biol. 2009, 212, 2075–2084. [Google Scholar] [CrossRef]
- Hickner, P.V.; Mori, A.; Zeng, E.; Tan, J.C.; Severson, D.W. Whole transcriptome responses among females of the filariasis and arbovirus vector mosquito Culex pipiens implicate TGF-β signaling and chromatin modification as key drivers of diapause induction. Funct. Integr. Genomics 2015, 15, 439–447. [Google Scholar] [CrossRef]
- Reynolds, J.A.; Hand, S.C. Decoupling development and energy flow during embryonic diapause in the cricket, Allonemobius socius. J. Exp. Biol. 2009, 212, 2065–2074. [Google Scholar] [CrossRef]
- Spielman, A.; Wong, J. Environmental control of ovarian diapause in Culex pipiens. Ann. Entomol. Soc. Am. 1973, 66, 905–907. [Google Scholar] [CrossRef]
- Clapier, C.R.; Cairns, B.R. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009, 78, 273–304. [Google Scholar] [CrossRef]
- Liu, M.; Pile, L.A. The transcriptional corepressor SIN3 directly regulates genes involved in methionine catabolism and affects histone methylation, linking epigenetics and metabolism. J. Biol. Chem. 2017, 292, 1970–1976. [Google Scholar] [CrossRef]
- Swaminathan, A.; Pile, L.A. Regulation of cell proliferation and wing development by Drosophila SIN3 and String. Mech. Dev. 2010, 127, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Barnes, V.L.; Bhat, A.; Unnikrishnan, A.; Heydari, A.R.; Arking, R.; Pile, L.A. SIN3 is critical for stress resistance and modulates adult lifespan. Aging 2014, 6, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Denlinger, D.L. Induction and termination of pupal diapause in Sarcophaga (Diptera: Sarcophagidae). Biol. Bull. 1972, 142, 11–24. [Google Scholar] [CrossRef]
- Huang, S.; Jiang, X.; Luo, L. Effects of photoperiod and temperature on diapause induction in the beet webworm Loxostege sticticalis Linnaeus (Lepidoptera: Pyralidae). Acta Entomol. Sin. 2009, 52, 274–280. [Google Scholar]
- Hahn, D.A.; Denlinger, D.L. Energetics of insect diapause. Annu. Rev. Entomol. 2011, 56, 103–121. [Google Scholar] [CrossRef]
- Sim, C.; Denlinger, D.L. Insulin signaling and the regulation of insect diapause. Front. Physiol. 2013, 4, 189–207. [Google Scholar] [CrossRef]
- Akhanaev, Y.B.; Berim, M.N.; Reznik, S.Y.; Saulich, A.K.; Frolov, A.N. On the temperature tolerance of diapausing prepupae of the beet webworm Loxostege sticticalis L. (Lepidoptera, Pyraloidea: Crambidae). Entomol. Rev. 2014, 94, 925–929. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- van Delft, J.H.M.; Mathijs, K.; Staal, Y.C.M.; van Herwijnen, M.H.M.; Brauers, K.J.J.; Boorsma, A.; Kleinjans, J.C.S. Time series analysis of benzo [A] pyrene-induced transcriptome changes suggests that a network of transcription factors regulates the effects on functional gene sets. Toxicol. Sci. 2010, 117, 381–392. [Google Scholar] [CrossRef] [PubMed]
- El-Sharkawy, I.; Liang, D.; Xu, K. Transcriptome analysis of an apple (Malus × domestica) yellow fruit somatic mutation identifies a gene network module highly associated with anthocyanin and epigenetic regulation. J. Exp. Bot. 2015, 66, 7359–7376. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Li, X.T.; Huang, Q.C.; Tang, Z.H. An review on the development of resistant mechanism in Chilo suppressalis. World Pestic. 2006, 28, 17–20. [Google Scholar]
- Denlinger, D.L. Diapause. Encyclopedia of Insects; Academic Press: Cambridge, MA, USA, 2009; pp. 267–271. [Google Scholar] [CrossRef]
- Saunders, D.S. Dormancy, diapause, and the role of the circadian system in insect photoperiodism. Annu. Rev. Entomol. 2020, 65, 373–389. [Google Scholar] [CrossRef]
- Bagchi, R.A.; Ferguson, B.S.; Stratton, M.S.; Hu, T.; Cavasin, M.A.; Sun, L.; Lin, Y.-H.; Liu, D.; Londono, P.; Song, K. HDAC11 suppresses the thermogenic program of adipose tissue via BRD2. JCI Insight 2018, 3, e120159. [Google Scholar] [CrossRef]
- Zha, L.; Li, F.; Wu, R.; Artinian, L.; Rehder, V.; Yu, L.; Liang, H.; Xue, B.; Shi, H. The histone demethylase UTX promotes brown adipocyte thermogenic program via coordinated regulation of H3K27 demethylation and acetylation. J. Biol. Chem. 2015, 290, 25151–25163. [Google Scholar] [CrossRef] [Green Version]
- Sim, C.; Kang, D.S.; Kim, S.; Bai, X.; Denlinger, D.L. Identification of FOXO targets that generate diverse features of the diapause phenotype in the mosquito Culex pipiens. Proc. Natl. Acad. Sci. USA 2015, 112, 3811–3816. [Google Scholar] [CrossRef]
- Latham, J.A.; Dent, S.Y.R. Cross-regulation of histone modifications. Nat. Struct. Mol. Biol. 2007, 14, 1017–1024. [Google Scholar] [CrossRef]
- Tissenbaum, H.A.; Guarente, L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 2001, 410, 227–230. [Google Scholar] [CrossRef]
- Qi, D.; Jin, H.; Lilja, T.; Mannervik, M. Drosophila Reptin and other TIP60 complex components promote generation of silent chromatin. Genetics 2006, 174, 241–251. [Google Scholar] [CrossRef]
- Santos, P.K.F.; de Souza Araujo, N.; Françoso, E.; Zuntini, A.R.; Arias, M.C. Diapause in a tropical oil-collecting bee: Molecular basis unveiled by RNA-Seq. BMC Genomics 2018, 19, 305. [Google Scholar] [CrossRef]
- George, S.; Gaddelapati, S.C.; Palli, S.R. Histone deacetylase 1 suppresses Krüppel homolog 1 gene expression and influences juvenile hormone action in Tribolium castaneum. Proc. Natl. Acad. Sci. USA 2019, 116, 17759–17764. [Google Scholar] [CrossRef]
- George, S.; Palli, S.R. Histone deacetylase 11 knockdown blocks larval development and metamorphosis in the red flour beetle, Tribolium castaneum. Front. Genet. 2020, 11, 683. [Google Scholar] [CrossRef]
- Lyu, H.; Xu, G.; Chen, P.; Song, Q.; Feng, Q.; Yi, Y.; Zheng, S. 20-Hydroxyecdysone receptor-activated Bombyx mori CCAAT/enhancer-binding protein gamma regulates the expression of BmCBP and subsequent histone H3 lysine 27 acetylation in Bo. mori. Insect Mol. Biol. 2020, 29, 256–270. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, J.; Lin, K.; Xu, L.; Yue, F.; Yu, L.; Zhang, Q. Transcriptome Analysis of Beet Webworm Shows That Histone Deacetylase May Affect Diapause by Regulating Juvenile Hormone. Insects 2022, 13, 835. https://doi.org/10.3390/insects13090835
Cui J, Lin K, Xu L, Yue F, Yu L, Zhang Q. Transcriptome Analysis of Beet Webworm Shows That Histone Deacetylase May Affect Diapause by Regulating Juvenile Hormone. Insects. 2022; 13(9):835. https://doi.org/10.3390/insects13090835
Chicago/Turabian StyleCui, Jin, Kejian Lin, Linbo Xu, Fangzheng Yue, Liangbin Yu, and Quanyi Zhang. 2022. "Transcriptome Analysis of Beet Webworm Shows That Histone Deacetylase May Affect Diapause by Regulating Juvenile Hormone" Insects 13, no. 9: 835. https://doi.org/10.3390/insects13090835