

Analysis of Polyadenylation Signal Usage with Full-Length Transcriptome in Spodoptera frugiperda (Lepidoptera: Noctuidae)

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. The Source of the Sequencing Data
2.2. Identification of Poly(A) Sites
2.3. Polyadenylation Signal Search and Analysis
2.4. APA Analysis in the Spodoptera frugiperda Genome
2.5. PAS Information Annotation
2.6. Data Analysis
3. Results
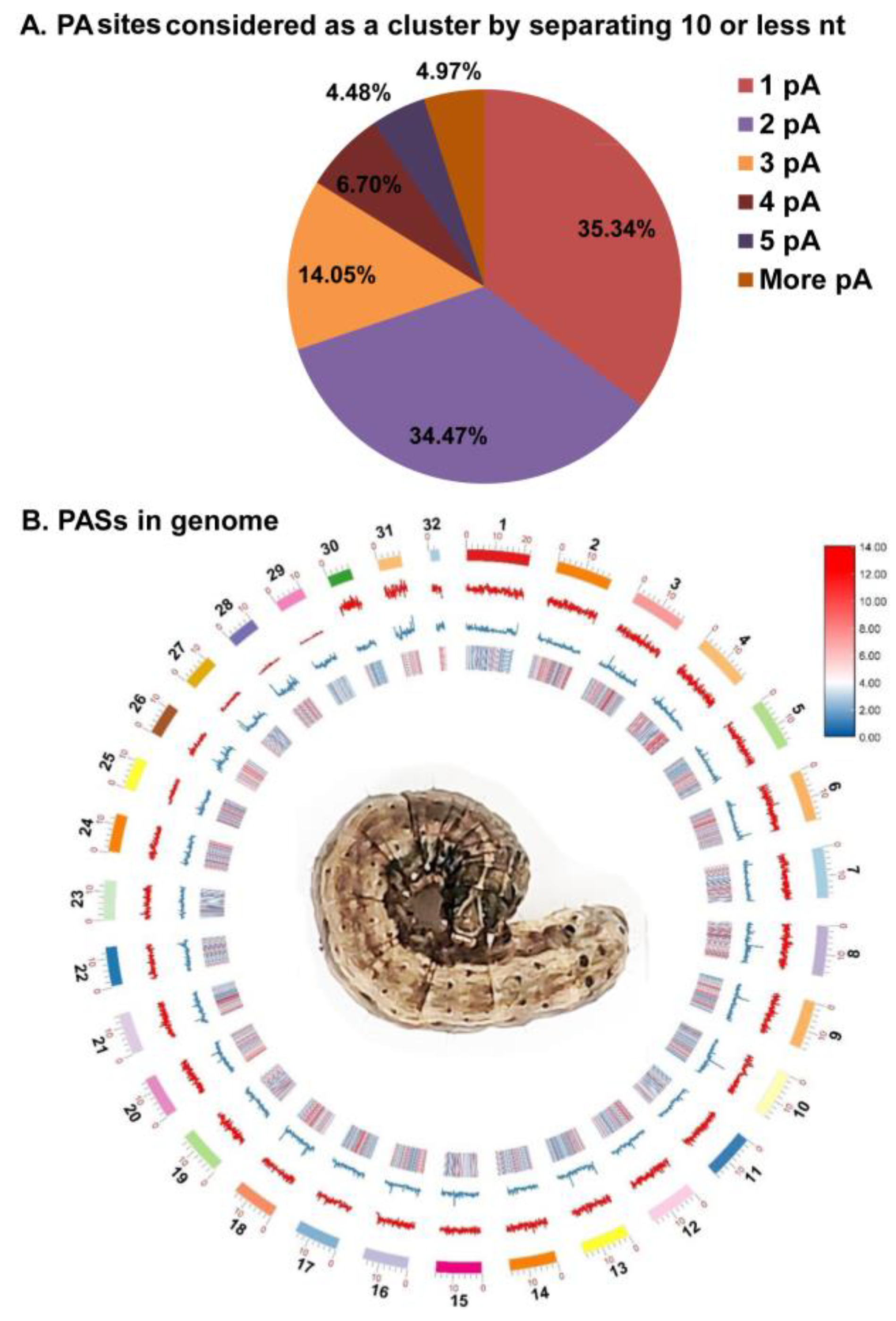
3.1. Identification of Polyadenylation Sites
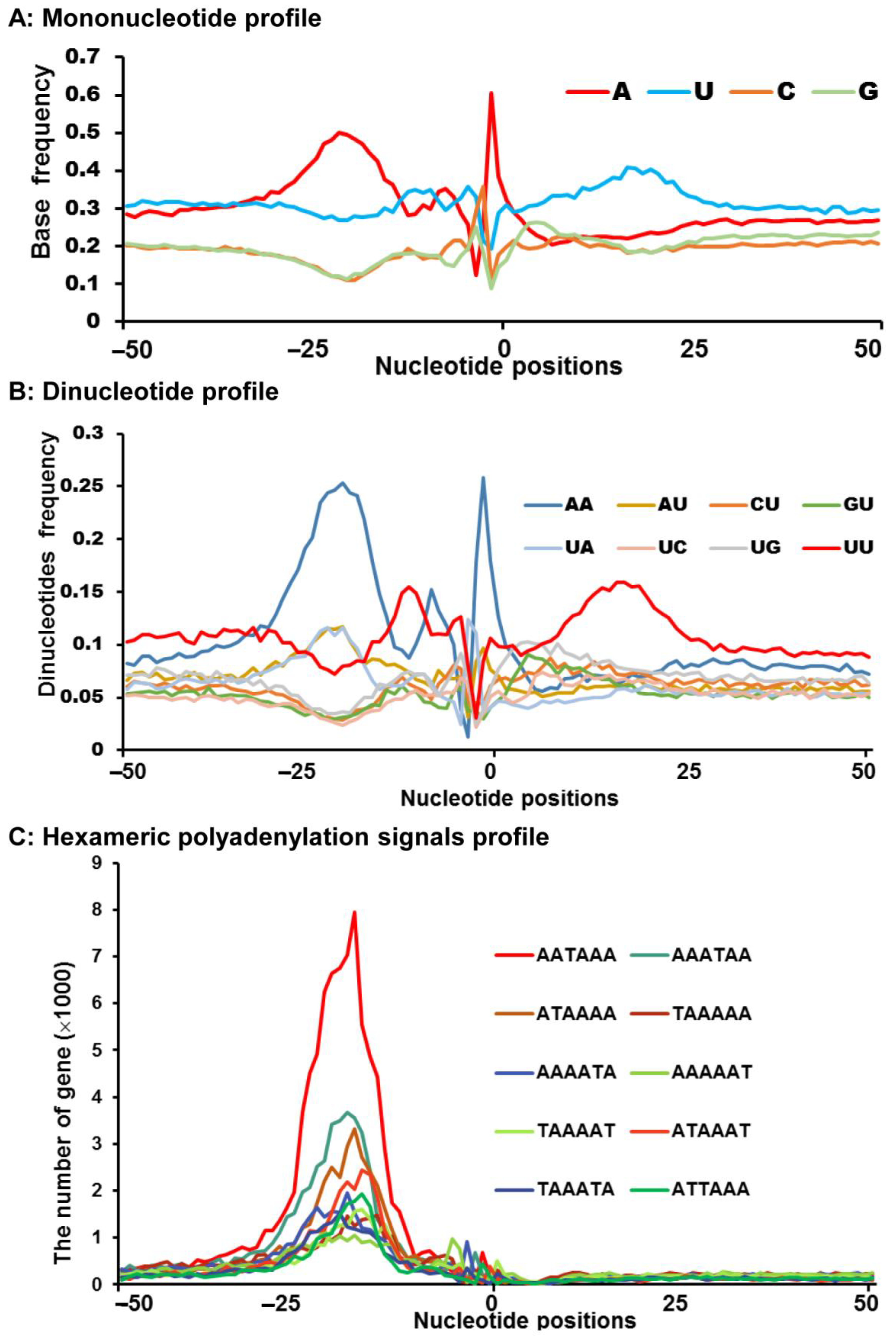
3.2. Nucleotide Bias Close to the Cleavage Sites
3.3. Sequence Motifs near the Cleavage Sites
3.4. Usage Frequencies of Core Hexamers Polyadenylation Signal
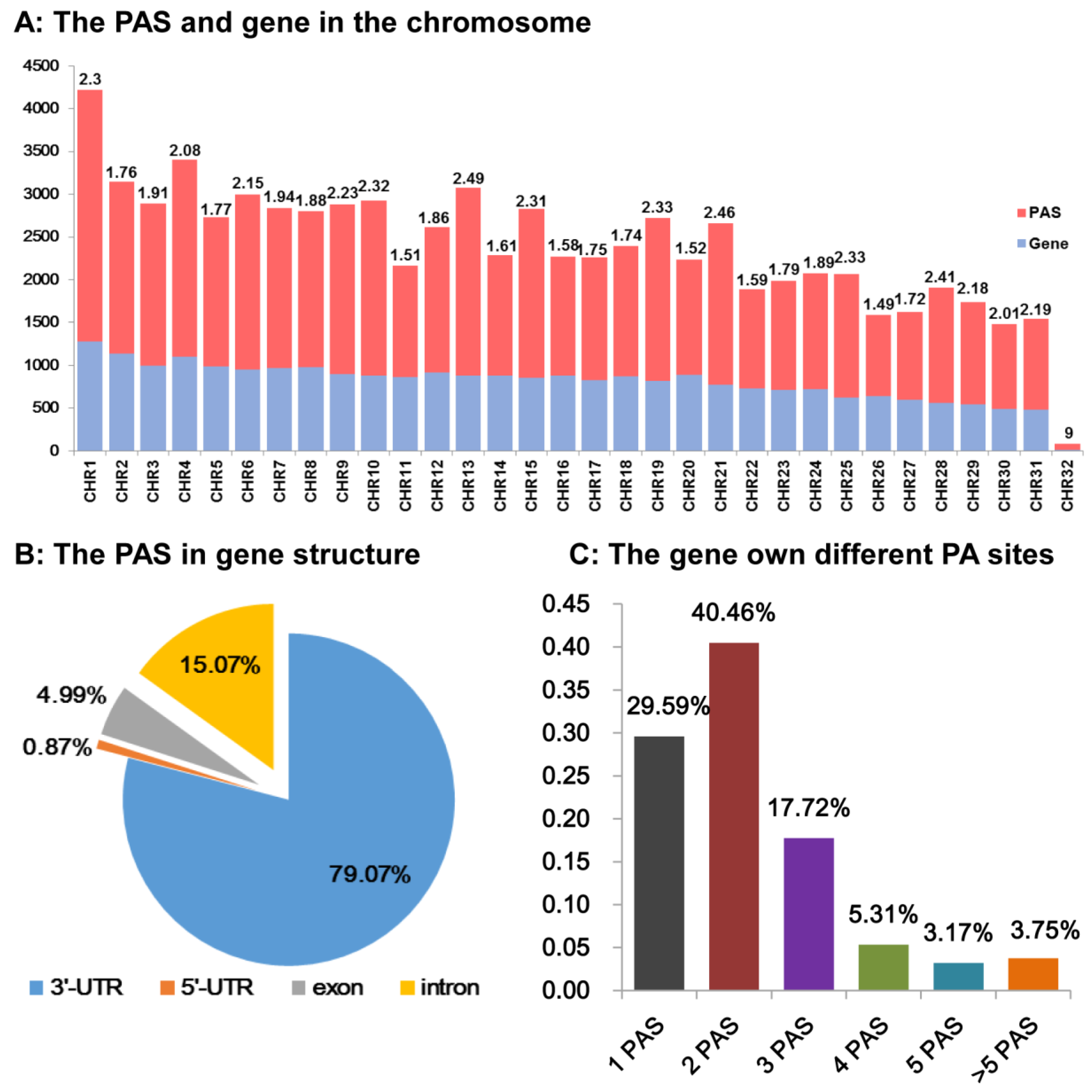
3.5. pA Sites Distribution in the Spodoptera frugiperda Genome
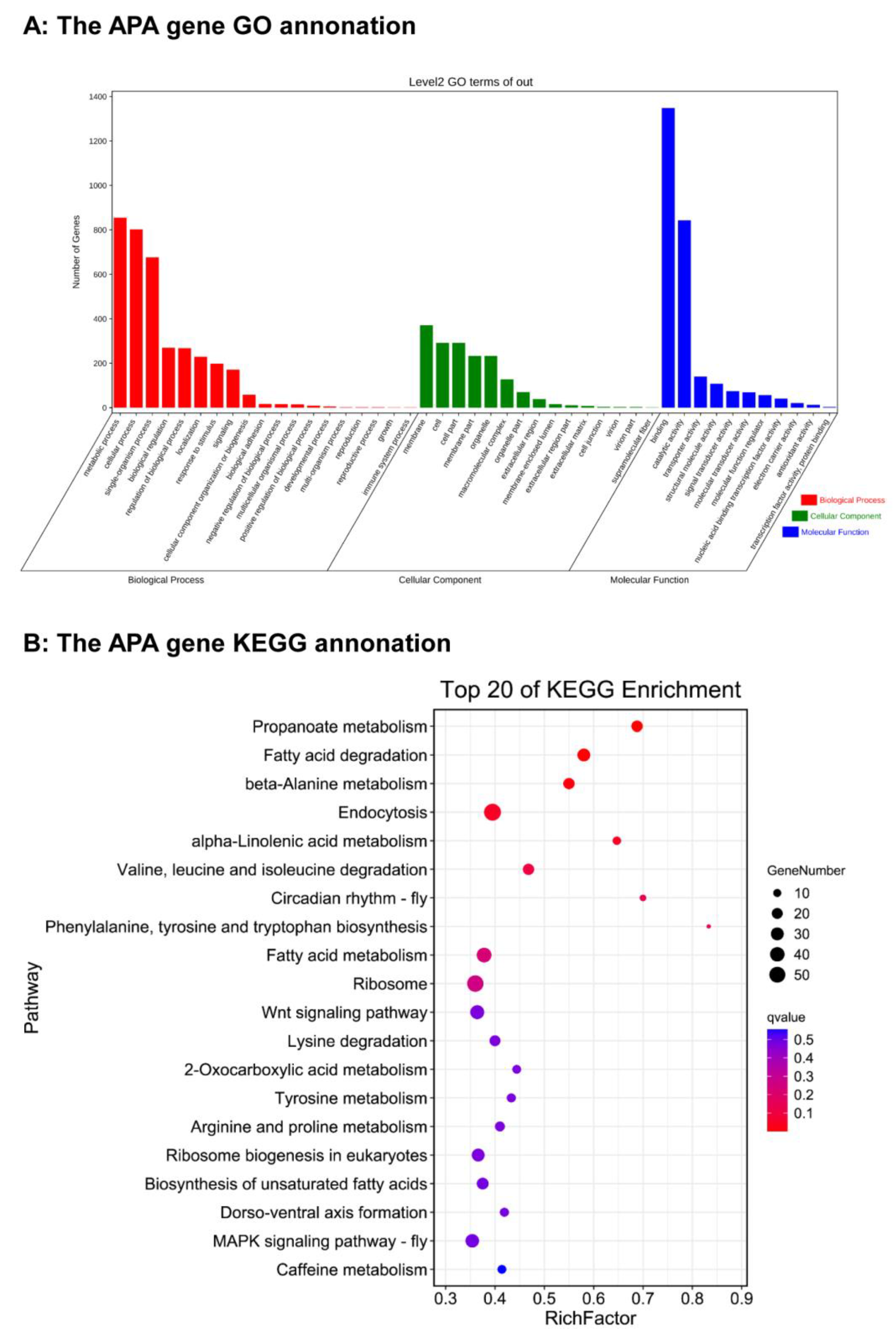
3.6. GO Analysis and KEGG Analysis of Alternative Polyadenylation Gene in Spodoptera frugiperda
3.7. Different Alternative Polyadenylation Types
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, Z.; Li, Y.; Krug, R.M. Influenza A virus NS1 protein targets poly(A)-binding protein II of the cellular 3′-end processing machinery. EMBO J. 1999, 18, 2273–2283. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Song, J.; Zhang, M.; Deng, Z. Analysis Polyadenylation Signal Usage in Sus scrofa. Animals 2022, 12, 194. [Google Scholar] [CrossRef] [PubMed]
- Jolles, B.; Jean-Jean, O. Poly(a) tail degradation in human cells: ATF4 mrna as a model for biphasic deadenylation. Biochimie 2021, 185, 128–134. [Google Scholar] [CrossRef]
- Xu, C.; Zhang, J. Alternative Polyadenylation of Mammalian Transcripts Is Generally Deleterious, Not Adaptive. Cell Syst. 2018, 6, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Libri, D. Endless Quarrels at the End of Genes. Mol. Cell 2015, 60, 192–194. [Google Scholar] [CrossRef]
- Elkon, R.; Ugalde, A.P.; Agami, R. Alternative cleavage and polyadenylation: Extent, regulation and function. Nat. Rev. Genet. 2013, 14, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jia, Q.; Song, Y.; Fu, H.; Wei, G.; Ni, T. Alternative polyadenylation:methods, findings, and impacts. J. Genom. Proteom. Bioinform. 2017, 15, 287–300. [Google Scholar] [CrossRef]
- Fitzgerald, M.; Shenk, T. The sequence 5′-AAUAAA-3′forms parts of the recognition site for polyadenylation of late SV40 mRNAs. Cell 1981, 24, 251–260. [Google Scholar] [CrossRef]
- Burri, D.; Zavolan, M. Shortening of 3′ UTRs in most cell types composing tumor tissues implicates alternative polyadenylation in protein metabolism. RNA 2021, 27, 1459–1470. [Google Scholar] [CrossRef]
- Beaudoing, E.; Freier, S.; Wyatt, J.R.; Claverie, J.M.; Gautheret, D. Patterns of variant polyadenylation signal usage in human genes. Genome Res. 2000, 10, 1001–1010. [Google Scholar] [CrossRef] [Green Version]
- Tian, B.; Hu, J.; Zhang, H.; Lutz, C.S. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 2005, 33, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Retelska, D.; Iseli, C.; Bucher, P.; Jongeneel, C.V.; Naef, F. Similarities and differences of polyadenylation signals in human and fly. BMC Genom. 2006, 7, 176. [Google Scholar] [CrossRef] [PubMed]
- Lakshmanan, V.; Bansal, D.; Kulkarni, J.; Poduval, D.; Krishna, S.; Sasidharan, V.; Anand, P.; Seshasayee, A.; Palakodeti, D. Genome-Wide Analysis of Polyadenylation Events in Schmidtea mediterranea. G3 2016, 6, 3035–3048. [Google Scholar] [CrossRef] [PubMed]
- Hoque, M.; Ji, Z.; Zheng, D.; Luo, W.; Li, W.; You, B. Analysis of alternative cleavage and polyadenylation by 3′ region extraction and deep sequencing. Nat. Methods 2013, 10, 133–139. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Y.; Shi, Y.; Walz, T.; Tong, L. Structural Insights into the Human Pre-mRNA 3′-End Processing Machinery. Mol. Cell 2019, 77, 800–809. [Google Scholar] [CrossRef]
- Shen, Y.; Ji, G.; Haas, B.J.; Wu, X.; Zheng, J.; Reese, G.J.; Li, Q.Q. Genome level analysis of rice mRNA 3′-end processing signals and alternative polyadenylation. Nucleic Acids Res. 2008, 36, 3150–3161. [Google Scholar] [CrossRef]
- Li, Y.; Schaefke, B.; Zou, X.; Zhang, M.; Heyd, F.; Sun, W.; Zhang, B.; Li, G.; Liang, W.; He, Y.; et al. Pan-tissue analysis of allelic alternative polyadenylation suggests widespread functional regulation. Mol. Syst. Biol. 2020, 16, e9367. [Google Scholar] [CrossRef]
- Guvenek, A.; Tian, B. Analysis of alternative cleavage and polyadenylation in mature and differentiating neurons using RNA-seq data. Quant. Biol. 2018, 6, 253–266. [Google Scholar] [CrossRef]
- Tian, B.; Manley, J.L. Alternative cleavage and polyadenylation: The long and short of it. Trends Biochem. Sci. 2013, 38, 312–320. [Google Scholar] [CrossRef]
- Neve, J.; Furger, A. Alternative polyadenylation: Less than meets the eye? Biochem. Soc. Trans. 2014, 42, 1190–1195. [Google Scholar] [CrossRef]
- Smibert, P.; Miura, P.; Westholm, J.O.; Shenker, S.; May, G.; Duff, M.O.; Zhang, D.; Eads, B.D.; Carlson, J.; Brown, J.B. Global patterns of tissue-specific alternative polyadenylation in Drosophila. Cell Rep. 2012, 1, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Chartier, A.; Joly, W.; Simonelig, M. Measurement of mRNA Poly(A) Tail Lengths in Drosophila Female Germ Cells and Germ-Line Stem Cells. Methods Mol. Biol. 2017, 1463, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Misof, B.; Liu, S.; Meusemann, K.; Peters, R.S.; Donath, A.; Mayer, C.; Frandsen, P.B.; Ware, J.; Flouri, T.; Beutel, R.G. Phylogenomics resolves the timing and pattern of insect evolution. Science 2014, 346, 763–767. [Google Scholar] [CrossRef]
- Montezano, D.G.; Specht, A.; Sosa-Gomez, D.R. Host plants of Spodoptera frugiperda (Lepidoptera: Noctuidae) in the Americas. Afr. Entomol. 2018, 26, 286–301. [Google Scholar] [CrossRef]
- FAO. Fall armyworm likely to spread from India to other parts of Asia with South East Asia and South China most at risk. In Rome: Food and Agriculture Organization of United Nations; FAO: Rome, Italy, 2018. [Google Scholar]
- Yang, X.; Liu, Y.; Luo, M.; Li, J.; Wang, W.; Wan, J.; Jiang, J. The first discovery of Spodoptera frugiperda in Jiangcheng County, Yunnan Province. Yunnan Agric. 2019, 1, 72. [Google Scholar]
- Kakumani, P.K.; Malhotra, P.; Mukherjee, S.K. A draft genome assembly of the army worm, Spodoptera frugiperda. Genomics 2014, 104, 134–143. [Google Scholar] [CrossRef]
- Gouin, A.; Bretaudeau, A.; Nam, K. Two genomes of highly polyphagous lepidopteran pests (Spodoptera frugiperda, Noctuidae) with different host-plant ranges. Sci. Rep. 2017, 7, 11816. [Google Scholar] [CrossRef]
- Nandakumar, S.; Ma, H.; Khan, A.S. Whole-genome sequence of the Spodoptera frugiperda Sf9 insect cell line. Microbiol. Resour. Announc. 2017, 5, e00829-17. [Google Scholar] [CrossRef]
- Liu, H.; Lan, T.; Fang, D.; Gui, F.; Wang, H.; Guo, W.; Chen, X.; Chang, Y.; He, S.; Lyu, L.; et al. Chromosome level draft genomes of the fall armyworm, Spodoptera frugiperda (Lepidoptera: Noctuidae), an alien invasive pest in China. BioRxiv 2019, 671560. [Google Scholar] [CrossRef]
- Xin, Y.; Yi, Y.; Yang, M.; Hua, X.; Fei, L.I. The genome annotation and comparative genomics analysis of spodoptera frugiperda. J. Environ. Entomol. 2019, 41, 706–717. [Google Scholar]
- Xiao, H.; Ye, X.; Xu, H.; Mei, Y.; Yang, Y.; Chen, X.; Yang, Y.; Liu, T.; Yu, Y.; Yang, W.; et al. The genetic adaptations of fall armyworm Spodoptera frugiperda facilitated its rapid global dispersal and invasion. Mol. Ecol. Resour. 2020, 20, 1050–1068. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, B.; Zheng, W.; Liu, C.; Zhang, D.; Zhao, S.; Li, Z.; Xu, P.; Wilson, K.; Withers, A.; et al. Genetic structure and insecticide resistance characteristics of fall armyworm populations invading China. Mol. Ecol. Resour. 2020, 20, 1682–1696. [Google Scholar] [CrossRef] [PubMed]
- Gimenez, S.; Abdelgaffar, H.; Goff, G.L.; Hilliou, F.; Blanco, C.A.; Hänniger, S.; Bretaudeau, A.; Fuentes, J.L.; Legeai, F. Adaptation by copy number variation increases insecticide resistance in the fall armyworm. Commun. Biol. 2020, 3, 664. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xing, B.; Li, F.; Wang, L.K.; Yuan, L.; Mbuji, A.L.; Peng, Z.; Malhat, F.; Wu, S. Full-length transcriptome analysis of Spodoptera frugiperda larval brain reveals detoxification genes. PeerJ 2021, 9, e12069. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, J.K.; Marioni, J.C.; Pai, A.A.; Degner, J.F.; Engelhardt, B.E.; Nkadori, E.; Veyrieras, J.B.; Stephens, M.; Gilad, Y.; Pritchard, J.K. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature 2010, 464, 768–772. [Google Scholar] [CrossRef]
- Arefeen, A.; Xiao, X.; Jiang, T. DeepPASTA: Deep neural network based polyadenylation site analysis. Bioinformatics 2019, 35, 4577–4585. [Google Scholar] [CrossRef]
- Jafar, Z.; Tariq, S.; Sadiq, I.; Nawaz, T.; Akhtar, M.N. Genome-Wide Profiling of Polyadenylation Events in Maize Using High-Throughput Transcriptomic Sequences. G3 2019, 9, 2749–2760. [Google Scholar] [CrossRef] [PubMed]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Types | EST | Full-Length Transcriptome |
|---|---|---|
| Raw reads | 65,423 | 7,510,007,630 |
| Poly(A) tail-containing reads | 35,853 | 375,500,381 |
| Reads mapped to the genome | 28,931 | 10,728,582 |
| pA sites | 10,245 | 40,371 |
| Total pA cluster | 50,616 | |
| Hexamers | Frequency (%) |
|---|---|
| AAUAAA | 51.64 |
| AUUAAA | 12.15 |
| UAUAAA | 4.38 |
| AGUAAA | 4.17 |
| AAGAAA | 2.63 |
| AAUAUA | 2.42 |
| AAUACA | 2.32 |
| CAUAAA | 2.00 |
| GAUAAA | 2.01 |
| AAUGAA | 1.64 |
| UUUAAA | 1.25 |
| ACUAAA | 1.19 |
| AAUAGA | 1.05 |
| AAAAAG | 1.03 |
| Other | 10.13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, L.; Guo, L.; Zhang, M.; Li, X.; Deng, Z. Analysis of Polyadenylation Signal Usage with Full-Length Transcriptome in Spodoptera frugiperda (Lepidoptera: Noctuidae). Insects 2022, 13, 803. https://doi.org/10.3390/insects13090803
Fang L, Guo L, Zhang M, Li X, Deng Z. Analysis of Polyadenylation Signal Usage with Full-Length Transcriptome in Spodoptera frugiperda (Lepidoptera: Noctuidae). Insects. 2022; 13(9):803. https://doi.org/10.3390/insects13090803
Chicago/Turabian StyleFang, Liying, Lina Guo, Min Zhang, Xianchun Li, and Zhongyuan Deng. 2022. "Analysis of Polyadenylation Signal Usage with Full-Length Transcriptome in Spodoptera frugiperda (Lepidoptera: Noctuidae)" Insects 13, no. 9: 803. https://doi.org/10.3390/insects13090803