
Population Genetic Structure and Population History of the Biting Midge Culicoides mahasarakhamense (Diptera: Ceratopogonidae)

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
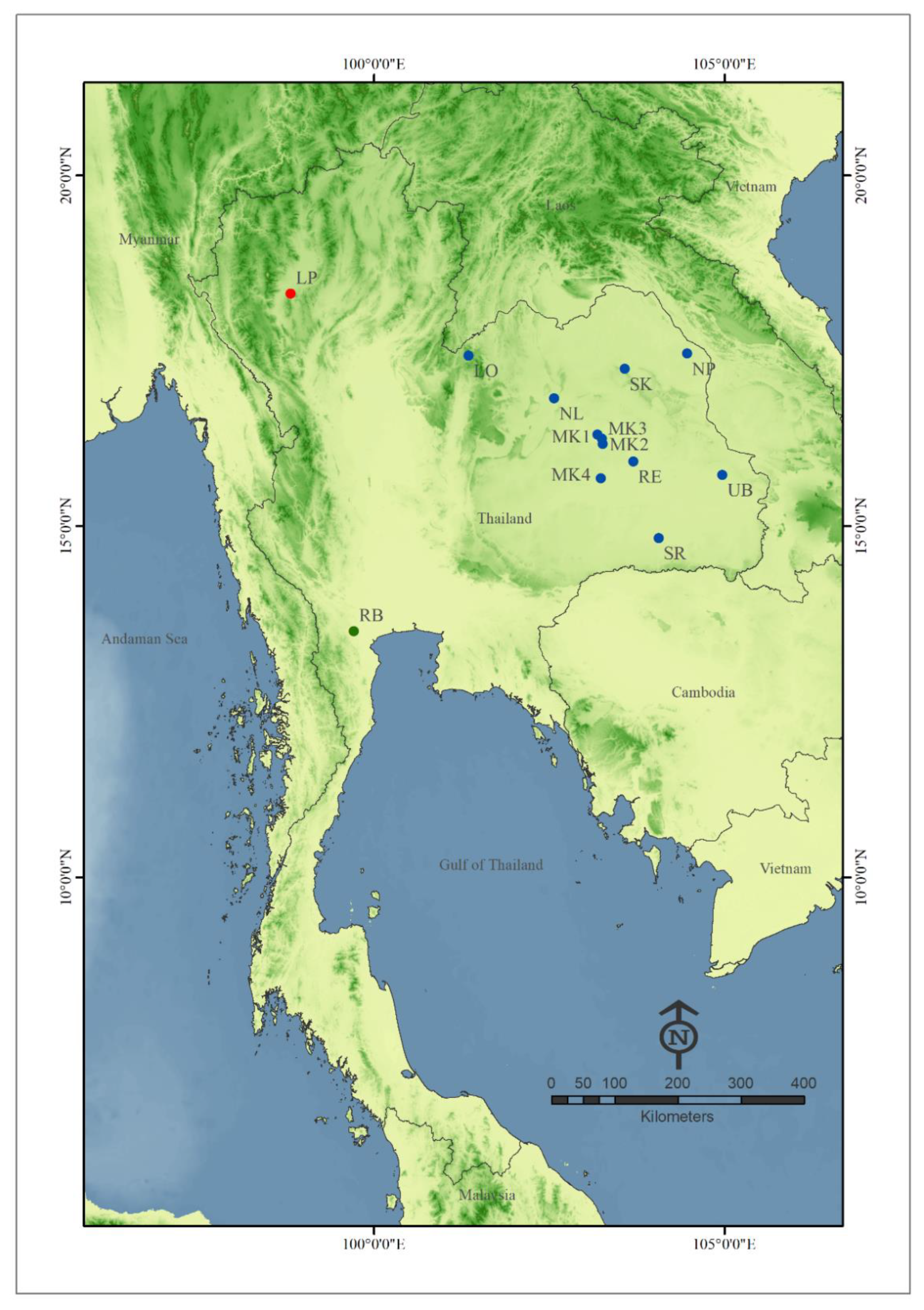
2.1. Specimen Collections and Identification
2.2. DNA Extraction, Polymerase Chain Reaction (PCR), and Sequencing
2.3. Data Analysis
3. Results
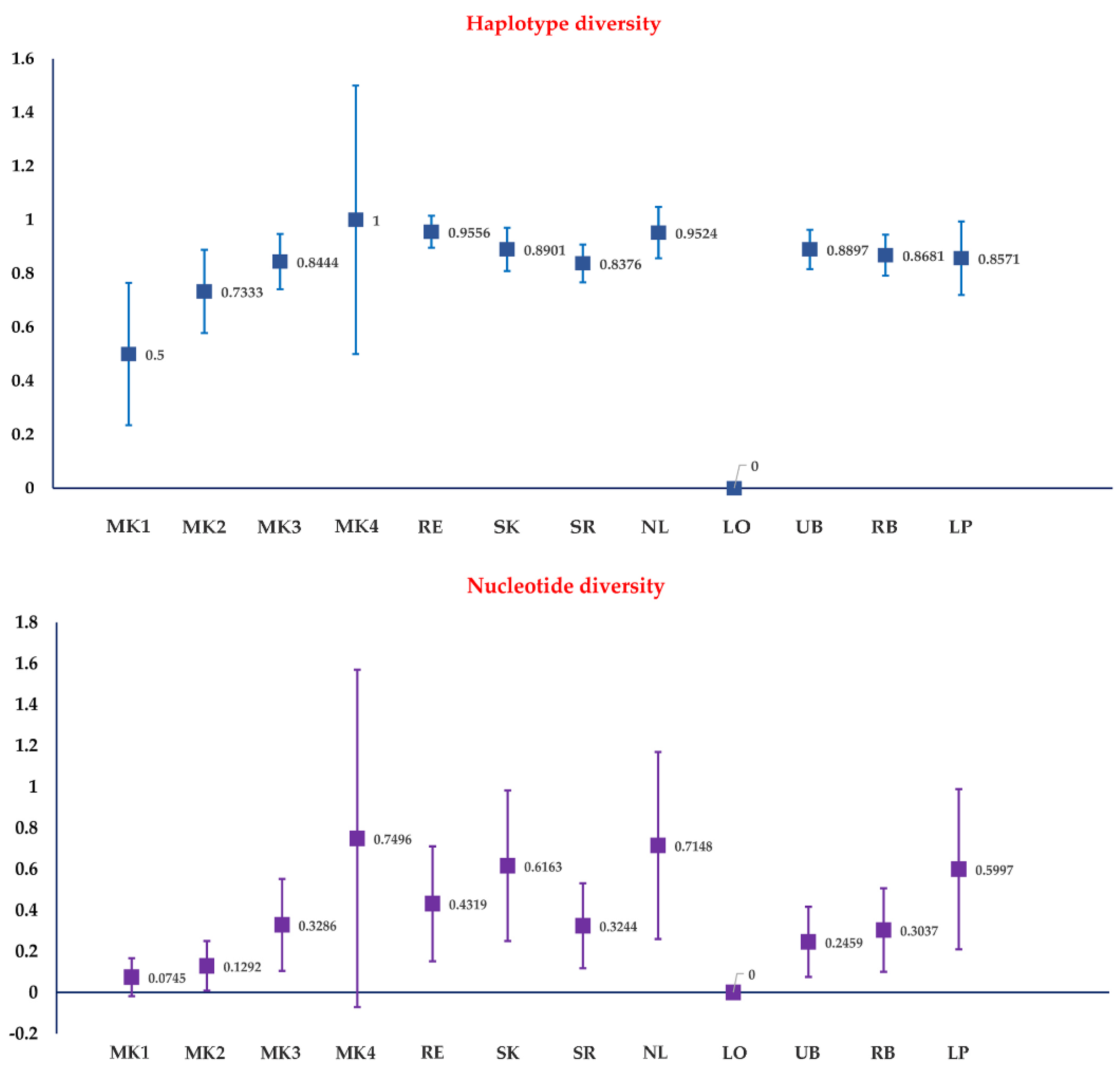
3.1. Genetic Diversity of Culicoides Mahasarakhamense
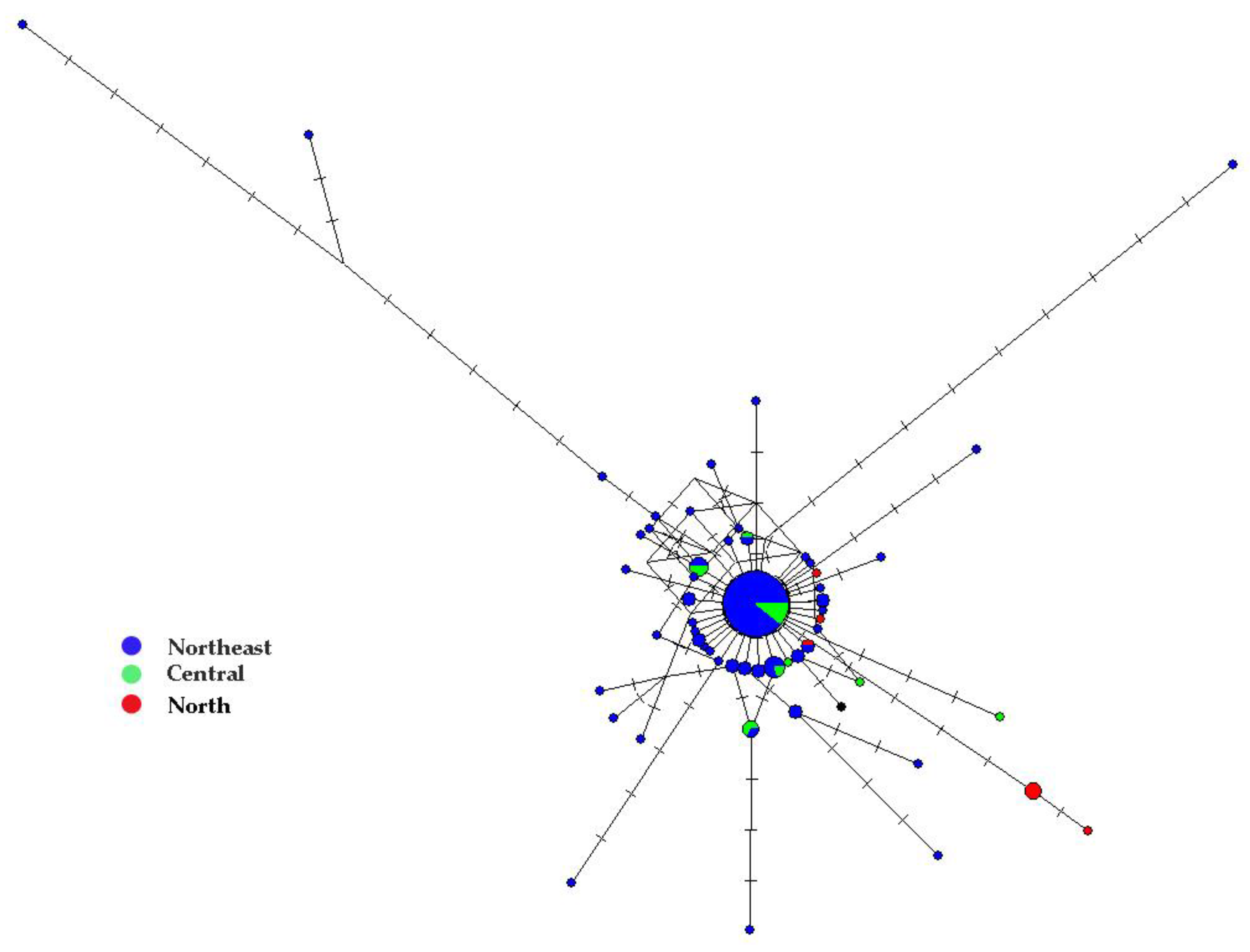
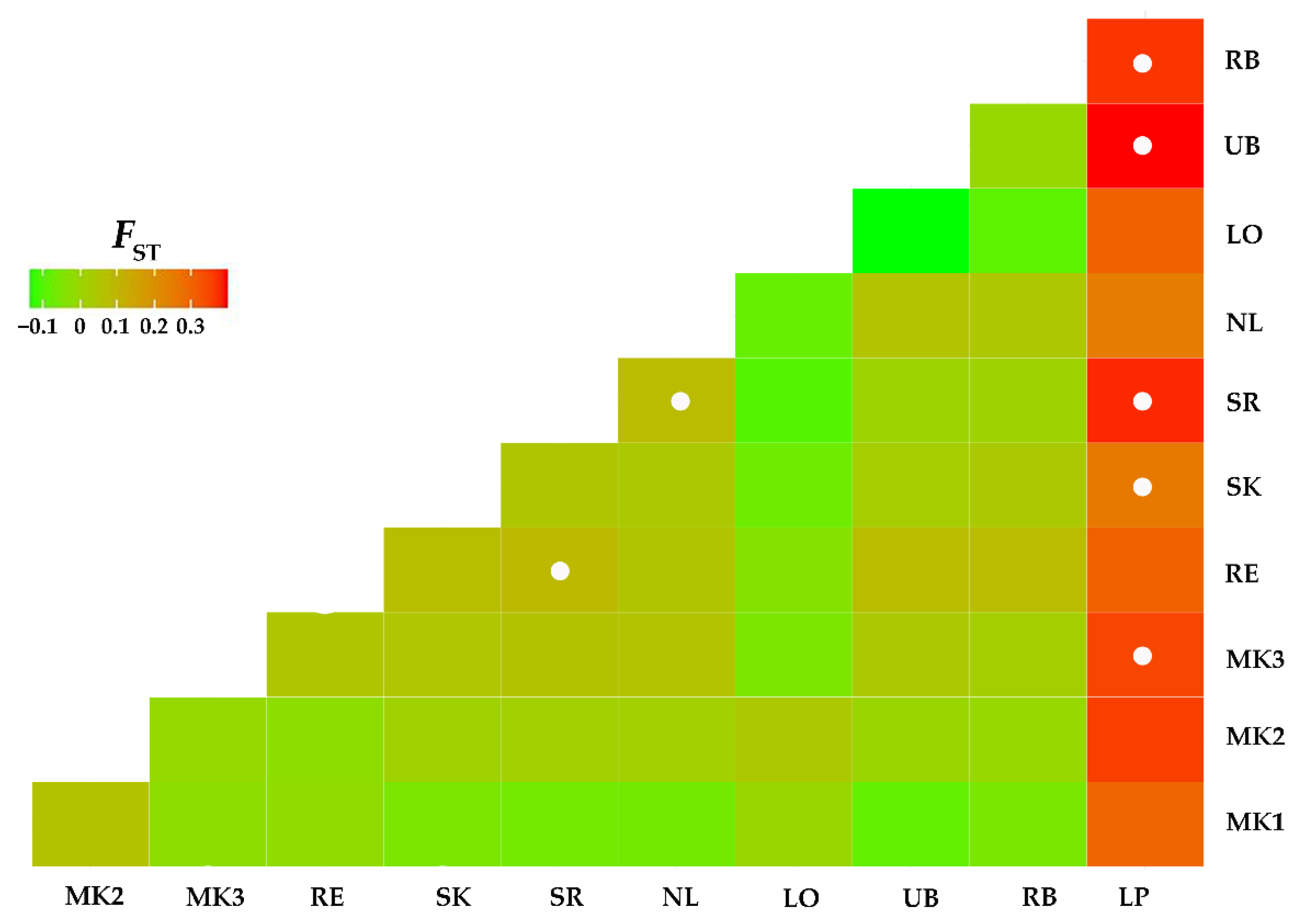
3.2. Mitochondrial Genealogy and Population Genetic Structure
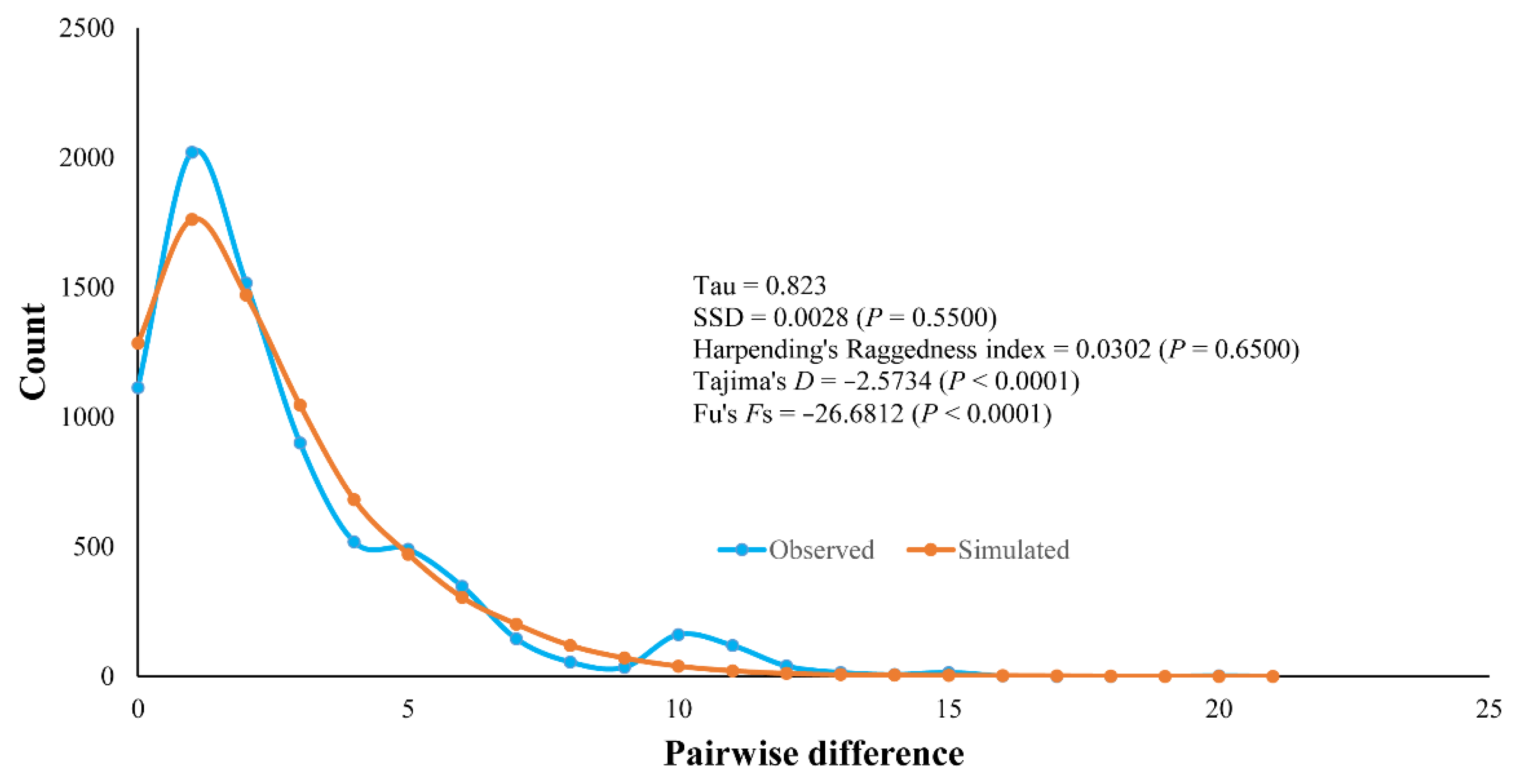
3.3. Population History
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Valkiūnas, G. Avian Malaria Parasites and Other Haemosporidia; CRC: Boca Raton, FL, USA, 2005. [Google Scholar]
- Santiago-Alarcon, D.; Palinauskas, V.; Schaefer, H.M. Diptera vectors of avian Haemosporidian parasites: Untangling parasite life cycles and their taxonomy. Biol. Rev. 2012, 87, 928–964. [Google Scholar] [CrossRef] [PubMed]
- Mullen, G.R.; Murphree, C.S. Biting midges (Ceratopogonidae). In Medical and Veterinary Entomology, 3rd ed.; Mullen, G.R., Durden, L.A., Eds.; Elsevier: San Diego, CA, USA, 2019; pp. 213–236. [Google Scholar]
- Tabachnick, W.J.; Black, I.V.W. Making a case for molecular population genetic studies of arthropod vectors. Parasitol. Today 1995, 11, 27–30. [Google Scholar] [CrossRef]
- Pérez De Rosas, A.R.; Segura, E.L.; Garcia, B.A. Microsatellite analysis of genetic structure in natural Triatoma infestans (Hemiptera: Reduviidae) populations from Argentina: Its implication in assessing the effectiveness of Chagas’ disease vector control programmes. Mol. Ecol. 2007, 16, 1401–1412. [Google Scholar] [CrossRef] [PubMed]
- McCoy, K.D. The population genetic structure of vectors and our understanding of disease epidemiology. Parasite 2008, 15, 444–448. [Google Scholar] [CrossRef]
- Vega-Rúa, A.; Marconcini, M.; Madec, Y.; Manni, M.; Carraretto, D.; Gomulski, L.M.; Gasperi, G.; Failloux, A.B.; Malacrida, A.R. Vector competence of Aedes albopictus populations for chikungunya virus is shaped by their demographic history. Commun. Biol. 2020, 3, 1–13. [Google Scholar] [CrossRef]
- Onyango, M.G.; Beebe, N.W.; Gopurenko, D.; Bellis, G.; Nicholas, A.; Ogugo, M.; Djikeng, A.; Kemp, S.; Walker, P.J.; Duchemin, J.B. Assessment of population genetic structure in the arbovirus vector midge, Culicoides brevitarsis (Diptera: Ceratopogonidae), using multi-locus DNA microsatellites. Vet. Res. 2015, 46, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, S.; Huber, K.; Talavera, S.; Burgin, L.E.; Carpenter, S.; Sanders, C.; Dicko, A.H.; Djerbal, M.; Goffredo, M.; Lhor, Y.; et al. Range expansion of the Bluetongue vector, Culicoides imicola, in continental France likely due to rare wind-transport events. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef]
- Calvo, J.H.; Calvete, C.; Martinez-Royo, A.; Estrada, R.; Miranda, M.A.; Borras, D.; Sato I Monteys, V.; Pages, N.; Delgado, J.A.; Collantes, F.; et al. Variations in the mitochondrial cytochrome c oxidase subunit I gene indicate northward expanding populations of Culicoides imicola in Spain. Bull. Entomol. Res. 2009, 99, 583–591. [Google Scholar] [CrossRef]
- Onyango, M.G.; Michuki, G.N.; Ogugo, M.; Venter, G.J.; Miranda, M.A.; Elissa, N.; Djikeng, A.; Kemp, S.; Walker, P.J.; Duchemin, J.B. Delineation of the population genetic structure of Culicoides imicola in East and South Africa. Parasit. Vectors 2015, 8, 1–13. [Google Scholar] [CrossRef]
- Jacquet, S.; Garros, C.; Lombaert, E.; Walton, C.; Restrepo, J.; Allene, X.; Baldet, T.; Cetre-Sossah, C.; Chaskopoulou, A.; Delecolle, J.C.; et al. Colonization of the Mediterranean basin by the vector biting midge species Culicoides imicola: An old story. Mol. Ecol. 2015, 24, 5707–5725. [Google Scholar] [CrossRef]
- Mignotte, A.; Garros, C.; Dellicour, S.; Jacquot, M.; Gilbert, M.; Gardès, L.; Balenghien, T.; Duhayon, M.; Rakotoarivony, I.; de Wavrechin, M.; et al. High dispersal capacity of Culicoides obsoletus (Diptera: Ceratopogonidae), vector of bluetongue and Schmallenberg viruses, revealed by landscape genetic analyses. Parasit. Vectors 2021, 14, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mellor, P.S.; Boorman, J.; Baylis, M. Culicoides biting midges: Their role as arbovirus vectors. Annu. Rev. Entomol. 2000, 45, 307–340. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, S.; Huber, K.; Guis, H.; Setier-Rio, M.L.; Goffredo, M.; Allène, X.; Rakotoarivony, I.; Chevillon, C.; Bouyer, J.; Baldet, T.; et al. Spatio-temporal genetic variation of the biting midge vector species Culicoides imicola (Ceratopogonidae) Kieffer in France. Parasit. Vectors 2016, 9, 1–12. [Google Scholar] [CrossRef]
- Pramual, P.; Jomkumsing, P.; Piraonapicha, K.; Jumpato, W. Integrative taxonomy uncovers a new Culicoides (Diptera: Ceratopogonidae) biting midge species from Thailand. Acta Trop. 2021, 220, 105941. [Google Scholar] [CrossRef] [PubMed]
- Pramual, P.; Jomkumsing, P.; Jumpato, W.; Bunauea, S. Molecular detection of avian haemosporidian parasites in biting midges (Diptera: Ceratopogonidae) from Thailand. Acta Trop. 2021, 224, 106118. [Google Scholar] [CrossRef]
- Sunantaraporn, S.; Thepparat, A.; Phumee, A.; Sor-Suwan, S.; Boonserm, R.; Bellis, G.; Siriyasatien, P. Culicoides Latreille (Diptera: Ceratopogonidae) as potential vectors for Leishmania martiniquensis and Trypanosoma sp. in northern Thailand. PLoS Negl. Trop. Dis. 2021, 15, e0010014. [Google Scholar] [CrossRef] [PubMed]
- Jomkumsing, P.; Surapinit, A.; Saengpara, T.; Pramual, P. Genetic variation, DNA barcoding and blood meal identification of Culicoides Latreille biting midges (Diptera: Ceratopogonidae) in Thailand. Acta Trop. 2021, 217, 105866. [Google Scholar] [CrossRef]
- Ruff, M.D. Important parasites in poultry production systems. Vet. Parasitol. 1999, 84, 337–347. [Google Scholar] [CrossRef]
- Pramual, P.; Kuvangkadilok, C.; Baimai, V.; Walton, C. Phylogeography of the black fly Simulium tani (Diptera: Simuliidae) from Thailand as inferred from mtDNA sequences. Mol. Ecol. 2005, 14, 3989–4001. [Google Scholar] [CrossRef]
- Chaiyasan, P.; Pramual, P. Population genetic structure and demographic history of the black fly vector, Simulium nodosum in Thailand. Med. Vet. Entomol. 2016, 30, 286–292. [Google Scholar] [CrossRef]
- O’loughlin, S.M.; Okabayashi, T.; Honda, M.; Kitazoe, Y.; Kishino, H.; Somboon, P.; Sochantha, T.; Nambanya, S.; Saikia, P.K.; Dev, V.; et al. Complex population history of two Anopheles dirus mosquito species in Southeast Asia suggests the influence of Pleistocene climate change rather than human-mediated effects. J. Evol. Biol. 2008, 21, 1555–1569. [Google Scholar] [CrossRef] [PubMed]
- Morgan, K.; O’Loughlin, S.M.; Chen, B.I.N.; Linton, Y.M.; Thongwat, D.; Somboon, P.; Fong, M.Y.; Butlin, R.; Verity, R.; Prakash, A.; et al. Comparative phylogeography reveals a shared impact of Pleistocene environmental change in shaping genetic diversity within nine Anopheles mosquito species across the Indo-Burma biodiversity hotspot. Mol. Ecol. 2011, 20, 4533–4549. [Google Scholar] [CrossRef] [PubMed]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar] [PubMed]
- Tangkawanit, U.; Wongpakam, K.; Pramual, P. A new black fly (Diptera: Simuliidae) species of the subgenus Asiosimulium Takaoka Choochote from Thailand. Zootaxa 2018, 4388, 111–122. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Meier, R.; Shiyang, K.; Vaidya, G.; Ng, P.K. DNA barcoding and taxonomy in Diptera: A tale of high intraspecific variability and low identification success. Syst. Biol. 2006, 55, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Mantel, N. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967, 27, 209–220. [Google Scholar] [PubMed]
- Bohonak, A.J. IBD (Isolation by Distance): A programme for analysis of isolation by distance. J. Hered. 2002, 93, 153–154. [Google Scholar] [CrossRef] [PubMed]
- Roger, A.R.; Harpending, H. Population growth makes waves in the distribution of pairwise genetic differences. Mol. Biol. Evol. 1992, 9, 552–569. [Google Scholar]
- Harpending, H.C. Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Hum. Biol. 1994, 66, 591–600. [Google Scholar] [PubMed]
- Papadopoulou, A.; Anastasiou, I.; Vogler, A.P. Revisiting the insect mitochondrial molecular clock: The mid-Aegean trench calibration. Mol. Biol. Evol. 2010, 27, 1659–1672. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.K. Laboratory colonization of biting midges (Diptera: Ceratopogonidae). J. Med. Entomol. 1974, 11, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Ander, M.; Troell, K.; Chirico, J. Barcoding of biting midges in the genus Culicoides: A tool for species determination. Med. Vet. Entomol. 2013, 27, 323–331. [Google Scholar] [CrossRef]
- Nielsen, S.A.; Kristensen, M. Delineation of Culicoides species by morphology and barcode exemplified by three new species of the subgenus Culicoides (Diptera: Ceratopogonidae) from Scandinavia. Parasit. Vectors 2015, 8, 151. [Google Scholar] [CrossRef] [PubMed]
- Harrup, L.E.; Laban, S.; Purse, B.V.; Reddy, Y.K.; Reddy, Y.N.; Byregowda, S.M.; Kumar, N.; Purushotham, M.; Kowali, S.; Prasad, M.; et al. DNA barcoding and surveillance sampling strategies for Culicoides biting midges (Diptera: Ceratopogonidae) in southern India. Parasit. Vectors 2016, 9, 461. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, M.T.; Sarr, M.; Fall, A.G.; Huber, K.; Fall, M.; Sembène, M.; Seck, M.T.; Labuschagne, K.; Gardès, L.; Ciss, M.; et al. DNA barcoding and molecular identification of field-collected Culicoides larvae in the Niayes area of Senegal. Parasit. Vectors 2018, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Brenner, R.J.; Wargo, M.J.; Stains, G.S.; Mulla, M.S. The dispersal of Culicoides mohave (Diptera: Ceratopogonidae) in the desert of southern California. Mosq. News 1984, 44, 343–350. [Google Scholar]
- Hendrickx, G.; Gilbert, M.; Staubach, C.; Elbers, A.; Mintiens, K.; Gerbier, G.; Ducheyne, E. A wind density model to quantify the airborne spread of Culicoides species during north-western Europe bluetongue epidemic, 2006. Prev. Vet. Med. 2008, 87, 162–181. [Google Scholar] [CrossRef] [PubMed]
- Ducheyne, E.; De Deken, R.; Bécu, S.; Codina, B.; Nomikou, K.; Mangana-Vougiaki, O.; Georgiev, G.; Purse, B.V.; Hendrickx, G. Quantifying the dispersal of Culicoides species in Greece and Bulgaria by wind. Geospat. Health 2007, 1, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Climate of Thailand. Available online: https://www.tmd.go.th/en/archive/thailand_climate.pdf (accessed on 3 August 2022).
- Purse, B.V.; Carpenter, S.; Venter, G.J.; Bellis, G.; Mullens, B.A. Bionomics of temperate and tropical Culicoides midges: Knowledge gaps and consequences for transmission of Culicoides-borne viruses. Annu. Rev. Entomol. 2015, 60, 373–392. [Google Scholar] [CrossRef] [PubMed]
- Avise, J.C. Phylogeography: The History and Formation of Species; Harvard University Press: Cambridge, MA, USA, 2000. [Google Scholar]
- Peters, J.; Lebrasseur, O.; Irving-Pease, E.K.; Paxinos, P.D.; Best, J.; Smallman, R.; Callou, C.; Gardeisen, A.; Trixl, S.; Frantz, L.; et al. The biocultural origins and dispersal of domestic chickens. Proc. Natl. Acad. Sci. USA 2022, 119, e2121978119. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location (Code) | Sampling Site Type | Region | N (hn) | Latitude/Longitude | Elevation (m) | Collection Date |
|---|---|---|---|---|---|---|
| 1. Ban Wai, Kantharawichai District, Maha Sarakham (MK1) | CS | Northeast | 4 (2) 1 | 16.3075° N/103.1891° E | 150 | 20 January 2021 |
| 2. Lerng Jan Reservior, Mueang Maha Sarakham District, Maha Sarakham Province (MK2) | CP | Northeast | 6 (3) 1 | 16.1730° N/103.2622° E | 140 | 28 January 2020 |
| 3. Mahasarakham University, Kantharawichai District, Maha Sarakham Province (MK3) | CP | Northeast | 10 (6) 1 | 16.2488° N/103.2505° E | 150 | 25 February 2019 |
| 4. Nadun District, Maha Sarakham Province (MK4) | CP/CS | Northeast | 2 (2) | 15.6830° N/103.2358° E | 140 | 20 February 2021 |
| 5. Ban Nong Bon, Mueang Roi Et District, Roi Et Province (RE) | CP/CS | Northeast | 10 (8) 1 | 15.9241° N/103.6980° E | 130 | 9 February 2020 |
| 6. Waritchaphum District, Sakon Nakhon Province (SK) | CP/CS | Northeast | 14 (10) | 17.2422° N/103.5744° E | 220 | 27 March 2021 |
| 7. Phon Sawan District, Nakhon Phanom Province (NP) | CP | Northeast | 1 (1) | 17.4616° N/104.4658° E | 150 | 20 November 2020 |
| 8. Prangku District, Sisaket Province (SR) | CP/CS | Northeast | 27 (15) | 14.8305° N/104.0605° E | 140 | 6 March 2021 |
| 9. Non Sang District, Nongbua Lampu Province (NL) | CP/CS | Northeast | 7 (6) | 16.8233° N/102.5688° E | 180 | 27 February 2021 |
| 10. Phu Ruea District, Loei Province (LO) | CS | Northeast | 4 (4) | 17.4308° N/101.3500° E | 650 | 20 January 2021 |
| 11. Kud Khaopun District, Ubon Ratchathani Province (UB) | CP/CS | Northeast | 17 (12) | 15.7330° N/104.9680° E | 130 | 13 January 2021 |
| 12. Paktho District, Ratchaburi Province (RB) | CP/CS | Central | 14 (8) | 13.5058° N/99.7125° E | 5 | 1 November 2021 |
| 13. Banhong District, Lamphun Province (LP) | N/A | North | 7 (5) 2 | 18.3169° N/98.8141° E | 300 | September 2019–January 2020 |
| Total | 123 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pramual, P.; Jomkumsing, P.; Wongpakam, K.; Vaisusuk, K.; Chatan, W.; Gomontean, B. Population Genetic Structure and Population History of the Biting Midge Culicoides mahasarakhamense (Diptera: Ceratopogonidae). Insects 2022, 13, 724. https://doi.org/10.3390/insects13080724
Pramual P, Jomkumsing P, Wongpakam K, Vaisusuk K, Chatan W, Gomontean B. Population Genetic Structure and Population History of the Biting Midge Culicoides mahasarakhamense (Diptera: Ceratopogonidae). Insects. 2022; 13(8):724. https://doi.org/10.3390/insects13080724
Chicago/Turabian StylePramual, Pairot, Panya Jomkumsing, Komgrit Wongpakam, Kotchaphon Vaisusuk, Wasupon Chatan, and Bhuvadol Gomontean. 2022. "Population Genetic Structure and Population History of the Biting Midge Culicoides mahasarakhamense (Diptera: Ceratopogonidae)" Insects 13, no. 8: 724. https://doi.org/10.3390/insects13080724