
Topical Fungal Infection Induces Shifts in the Gut Microbiota Structure of Brown Planthopper, Nilaparvata lugens (Homoptera: Delphacidae)

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Insect and Fungal Entomopathogen
2.2. Topical Fungal Infection and Gut Dissection
2.3. Quantification of Gut Bacteria by CFU Counting Assay
2.4. Quantification of Gut Bacteria by 16S rRNA Gene qPCR Assay
2.5. DNA Extraction, PCR Amplification and High-Throughput Sequencing
2.6. 16S rRNA Gene Amplicon Sequence Analysis
2.7. qRT-PCR Analysis of Gut-Homeostasis-Related Genes
2.8. Statistical Analysis
3. Results
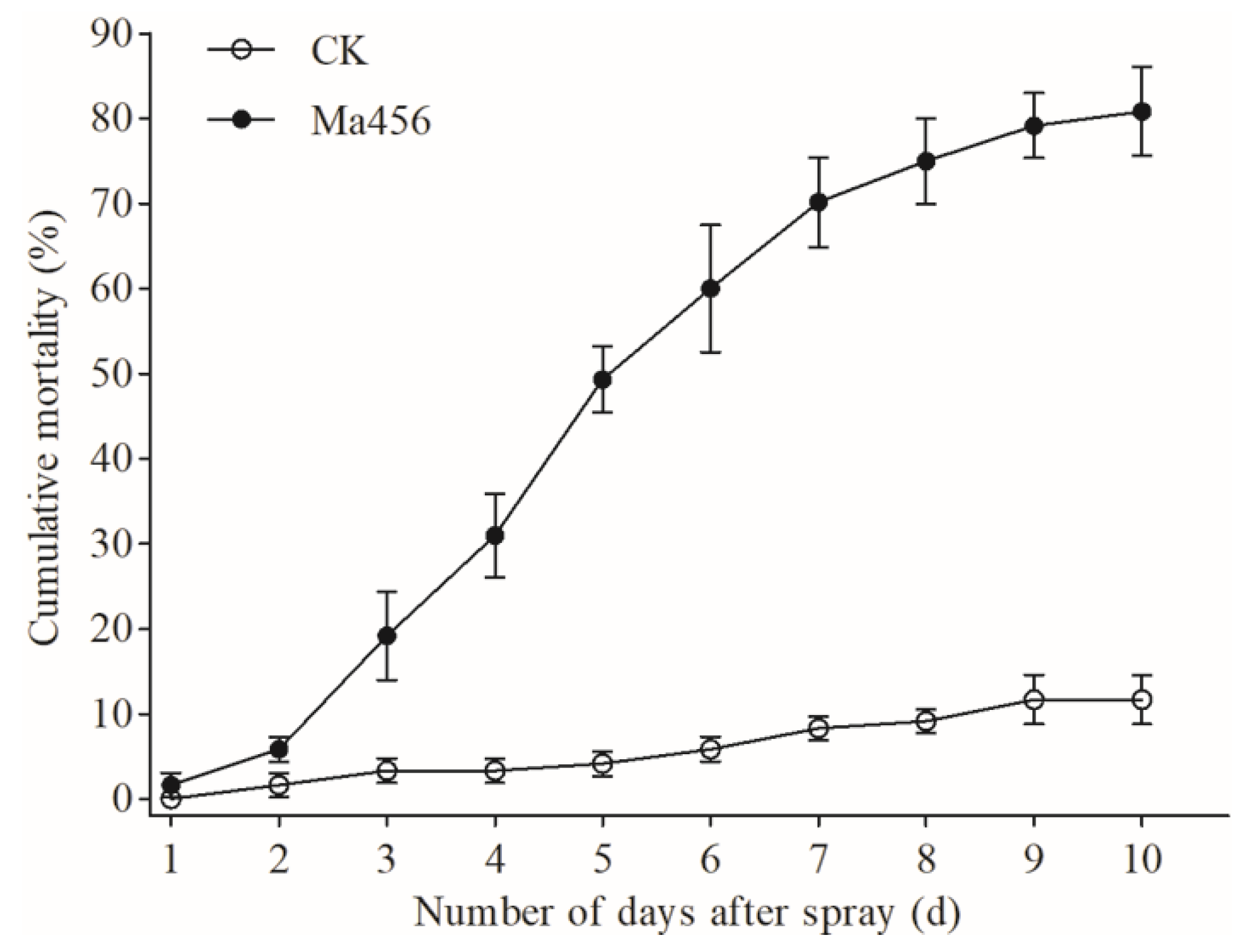
3.1. Fungal Infection Caused High Mortality of BPH Nymphs
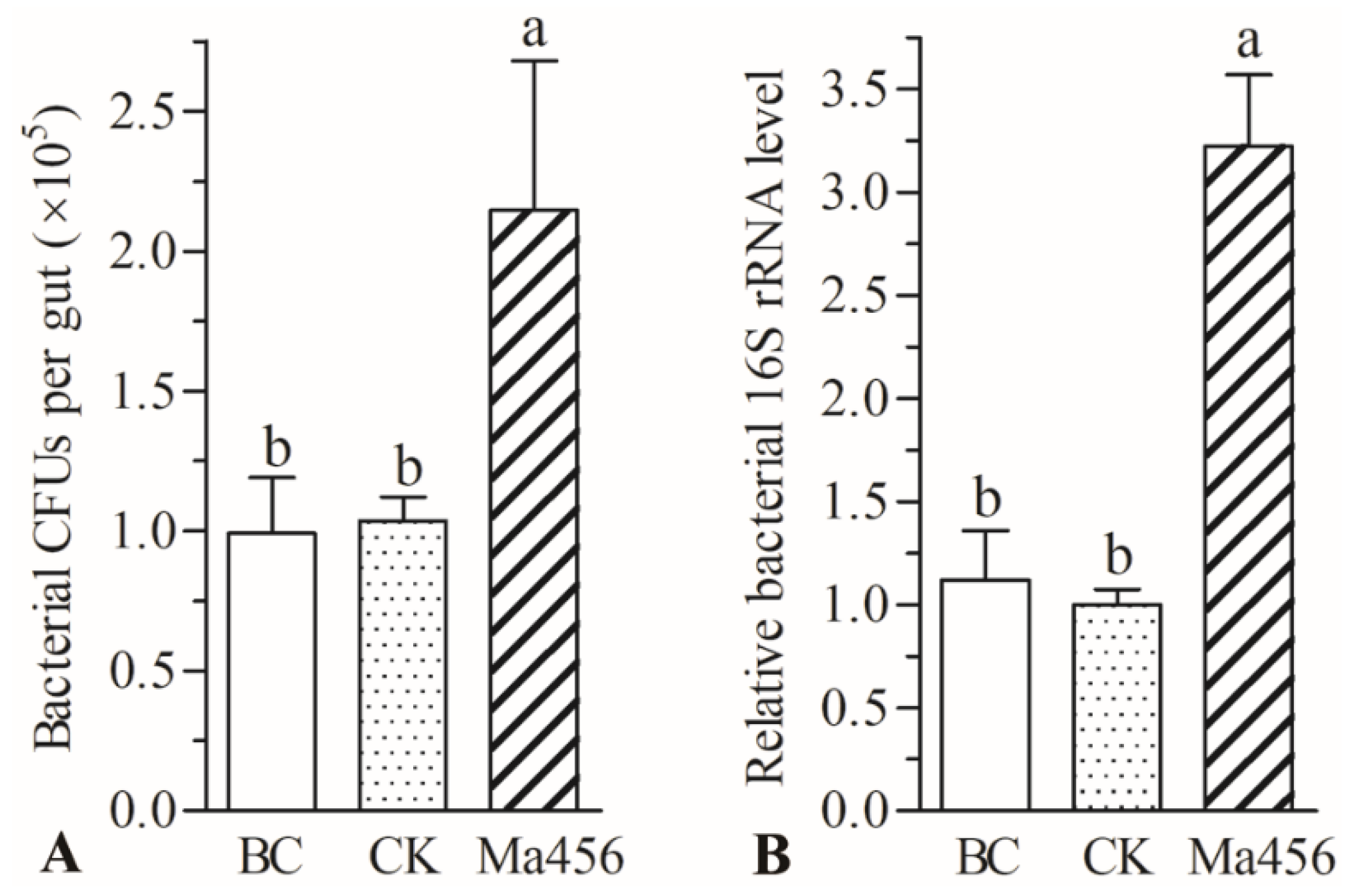
3.2. Fungal Infection Enhanced Bacterial Load in BPH Gut
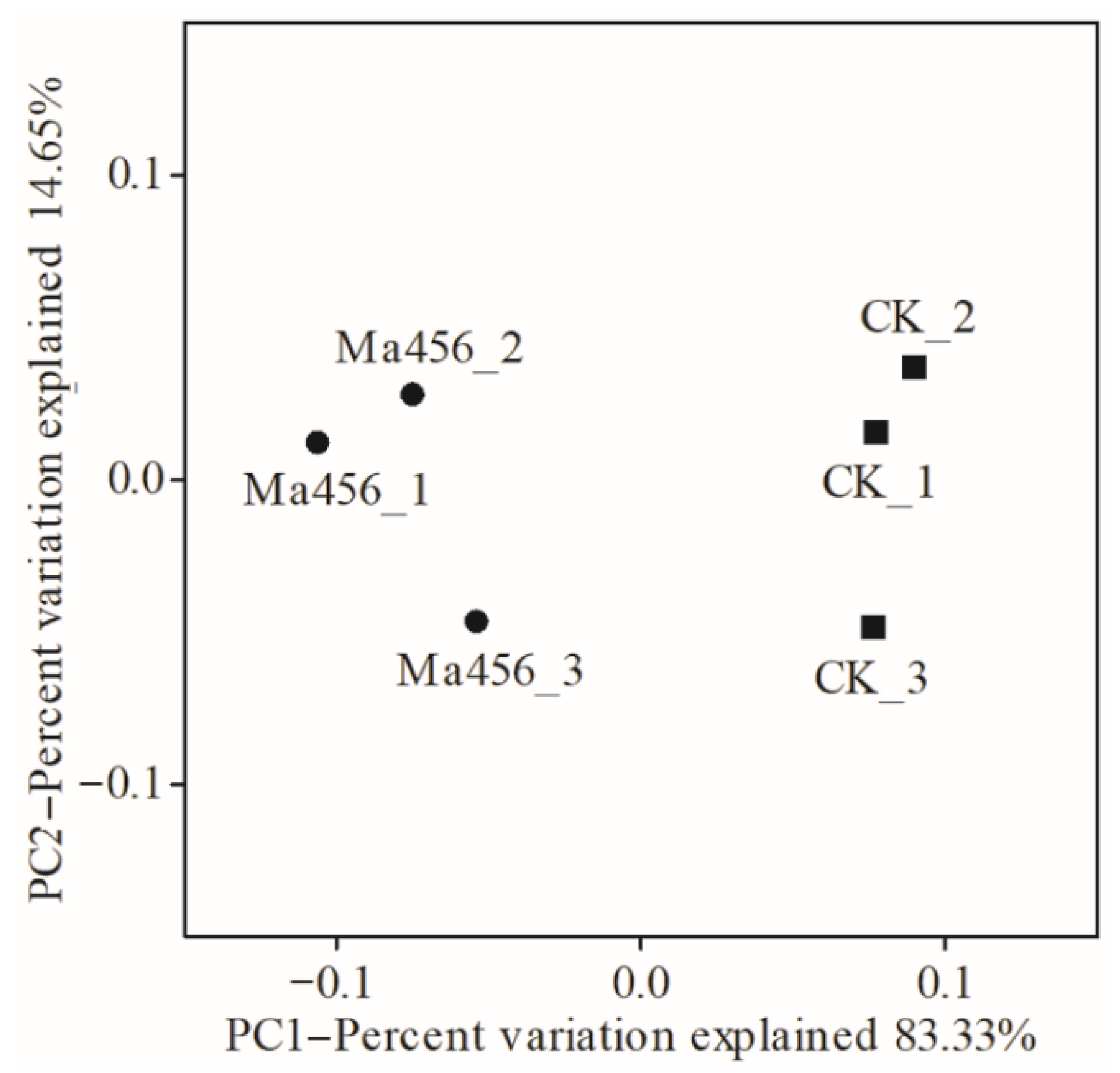
3.3. Fungal Infection Decreased Bacterial Community Evenness in BPH Gut
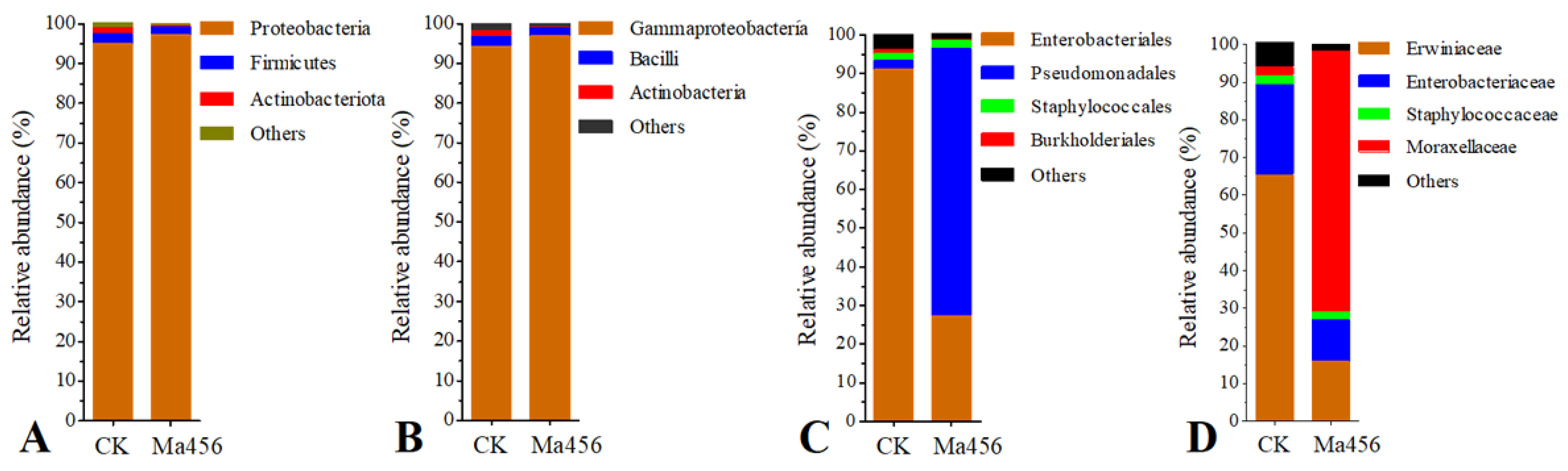
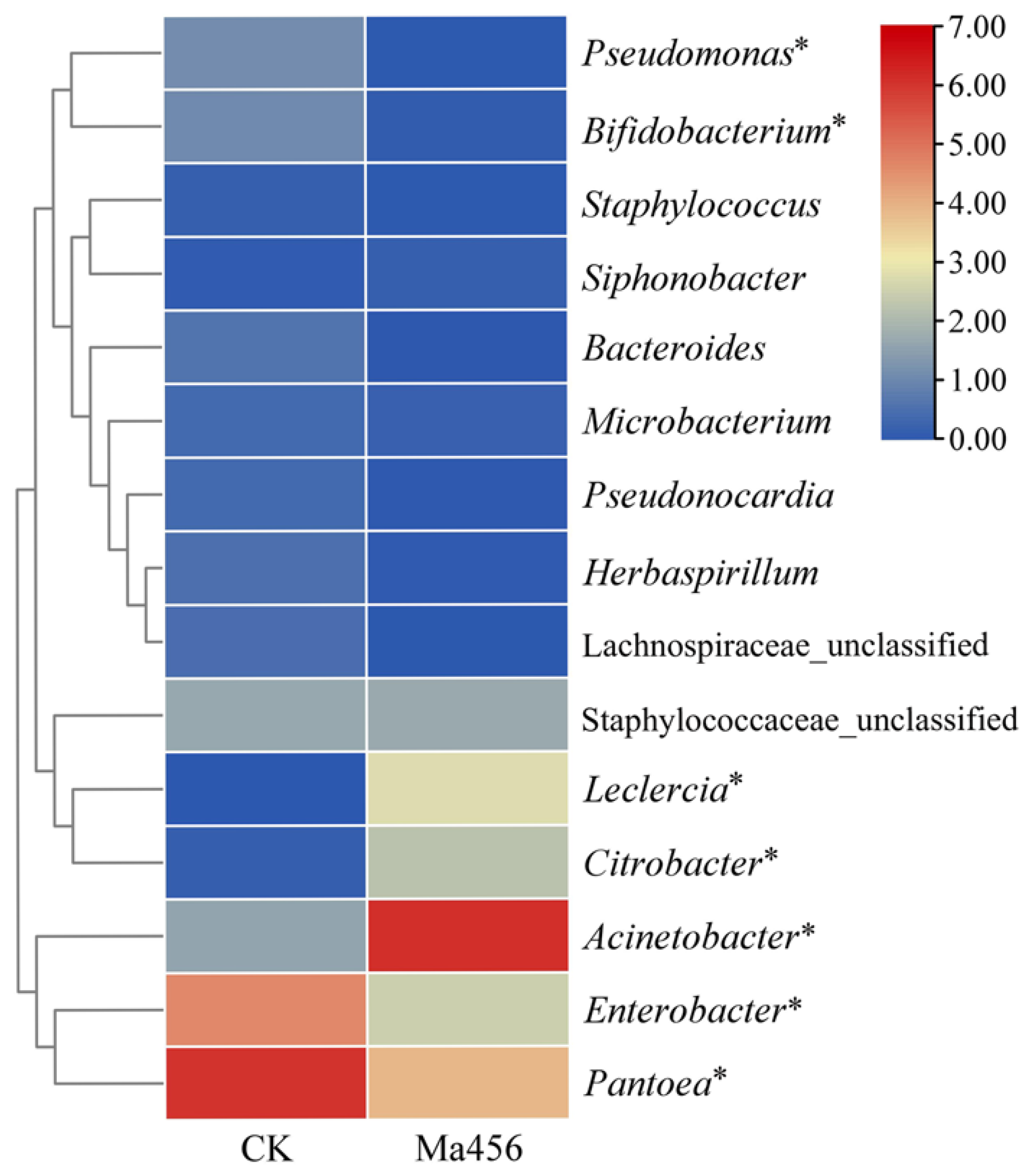
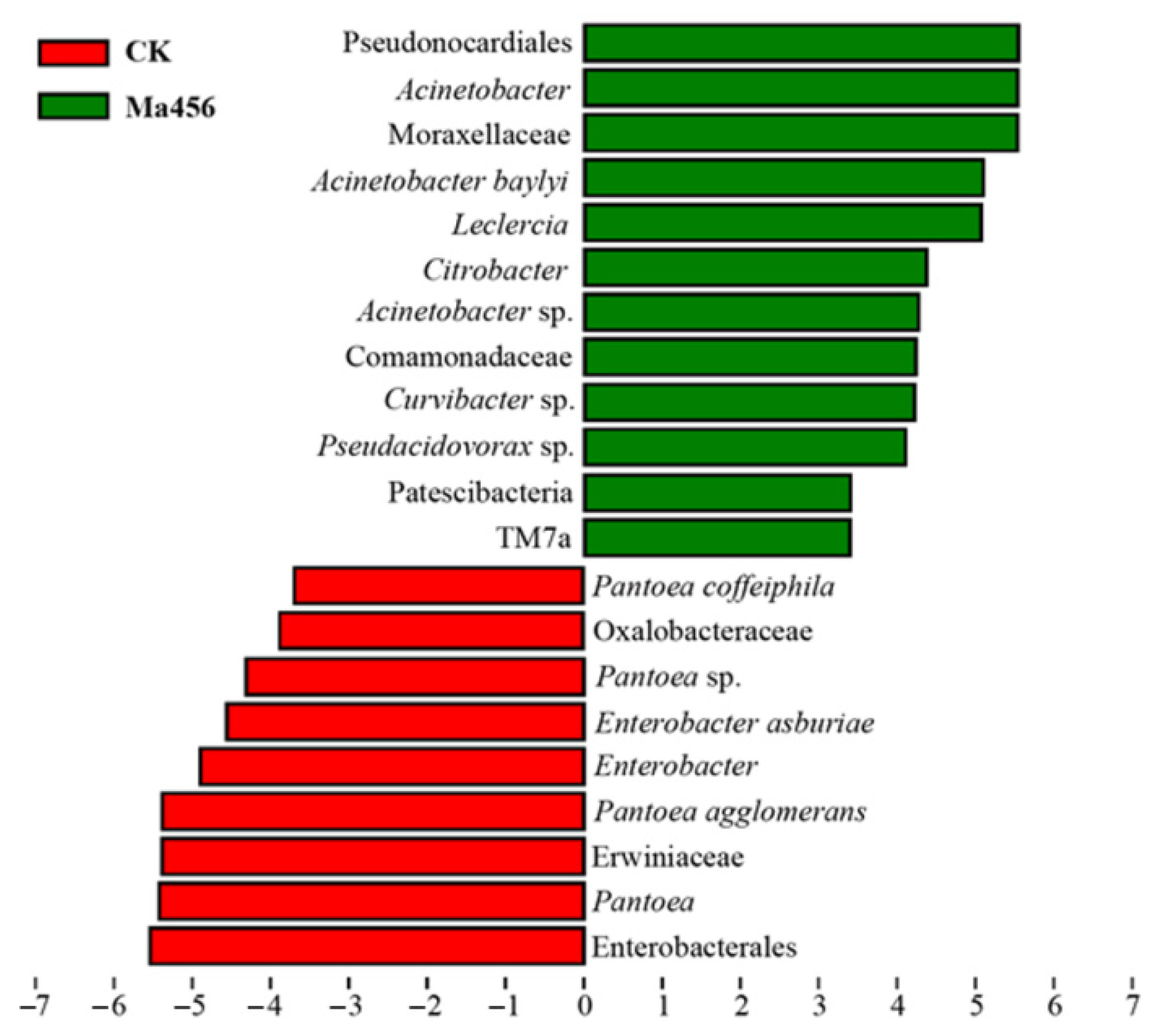
3.4. Fungal Infection Altered Bacterial Community Composition in BPH Gut
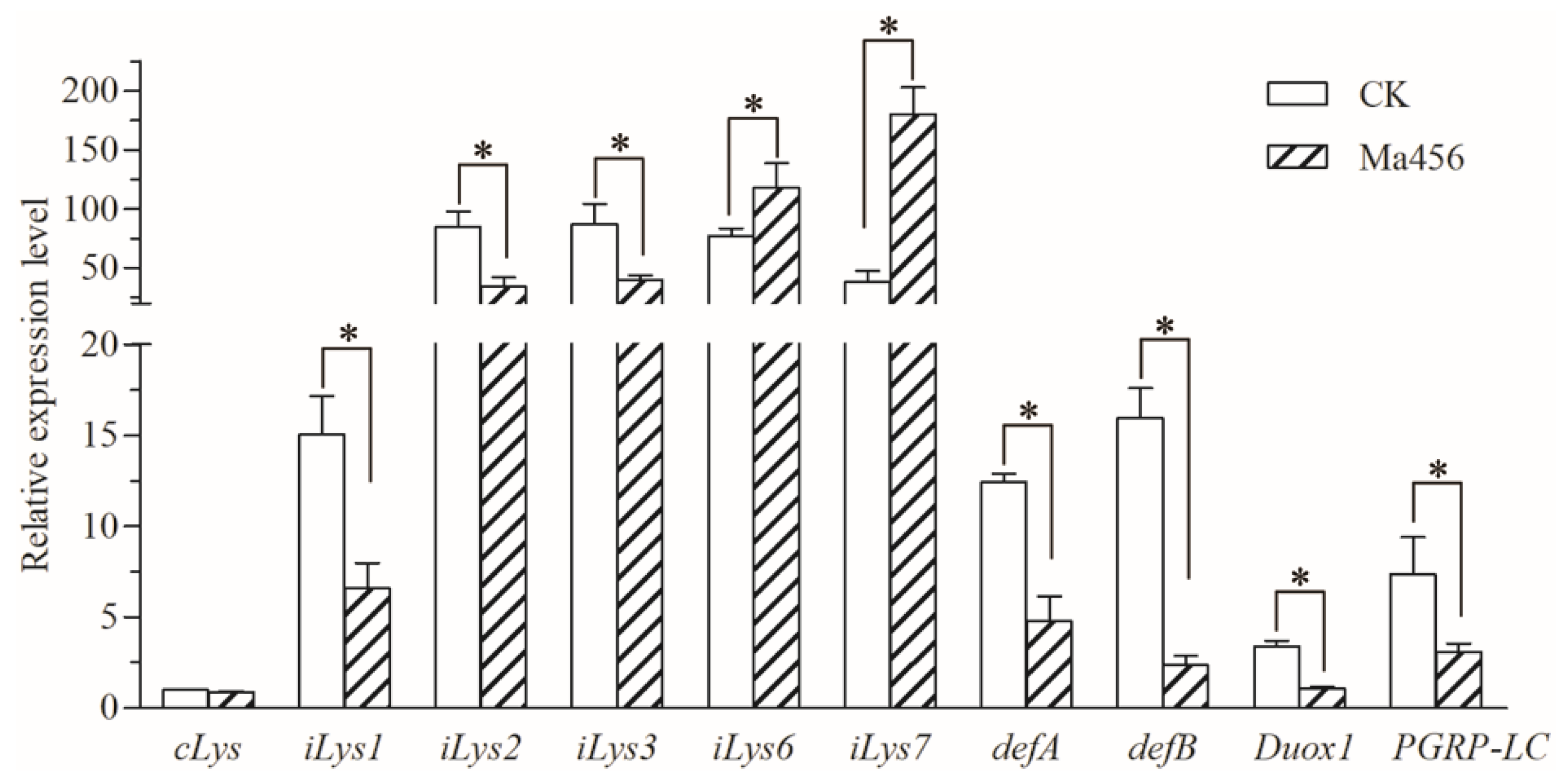
3.5. Fungal Infection Modulated Expressions of Gut-Homeostasis-Related Genes in BPH
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cheng, X.; Zhu, L.; He, G. Towards understanding of molecular interactions between rice and the brown planthopper. Mol. Plant 2013, 6, 621–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jena, K.K.; Kim, S.M. Current status of brown planthopper (BPH) resistance and genetics. Rice 2010, 3, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.F.; Feng, M.G.; Ying, S.H.; Mu, W.J.; Chen, J.Q. Evaluation of alternative rice planthopper control by the combined action of oil-formulated Metarhizium anisopliae and low-rate buprofezin. Pest Manag. Sci. 2011, 67, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.F.; Liu, X.Y.; Ding, Y.C.; Jiang, W.J.; Xie, J.Q. Evaluation of Metarhizium anisopliae for rice planthopper control and its synergy with selected insecticides. Crop Prot. 2019, 121, 132–138. [Google Scholar] [CrossRef]
- Zhao, Q.; Ye, L.; Wang, Z.; Li, Y.; Zhang, Y.; Keyhani, N.O.; Huang, Z. Sustainable control of the rice pest, Nilaparvata lugens, using the entomopathogenic fungus Isaria javanica. Pest Manag. Sci. 2021, 77, 1452–1464. [Google Scholar] [CrossRef]
- Li, M.; Li, S.; Xu, A.; Lin, H.; Chen, D.; Wang, H. Selection of Beauveria isolates pathogenic to adults of Nilaparvata lugens. J. Insect Sci. 2014, 14, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Peng, G.; Xie, J.; Guo, R. Long-term field evaluation and large-scale application of a Metarhizium anisopliae strain for controlling major rice pests. J. Pest Sci. 2021, 94, 969–980. [Google Scholar] [CrossRef]
- Islam, W.; Adnan, M.; Shabbir, A.; Naveed, H.; Abubakar, Y.S.; Qasim, M.; Tayyab, M.; Noman, A.; Nisar, M.S.; Khan, K.A.; et al. Insect-fungal-interactions: A detailed review on entomopathogenic fungi pathogenicity to combat insect pests. Microb. Pathog. 2021, 159, 105122. [Google Scholar] [CrossRef]
- Lu, H.L.; St Leger, R.J. Insect immunity to entomopathogenic fungi. Adv. Genet. 2016, 94, 251–285. [Google Scholar]
- Vertyporokh, L.; Wojda, I. Expression of the insect metalloproteinase inhibitor IMPI in the fat body of Galleria mellonella exposed to infection with Beauveria bassiana. Acta Biochim. Pol. 2017, 64, 273–278. [Google Scholar] [CrossRef]
- Chakraborty, M.; Mandal, S.M.; Basak, A.; Ghosh, A.K. Identification of a novel humoral antifungal defense molecule in the hemolymph of tasar silkworm Antheraea mylitta. Biochem. Biophys. Res. Commun. 2019, 519, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Al Souhail, Q.; Hiromasa, Y.; Rahnamaeian, M.; Giraldo, M.C.; Takahashi, D.; Valent, B.; Vilcinskas, A.; Kanost, M.R. Characterization and regulation of expression of an antifungal peptide from hemolymph of an insect, Manduca sexta. Dev. Comp. Immunol. 2016, 61, 258–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Tang, J.; Xie, J. Transcriptomic analysis of the brown planthopper, Nilaparvata lugens, at different stages after Metarhizium anisopliae challenge. Insects 2020, 11, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.L.; Pan, H.B.; Li, M.Y.; Wu, W.; Yu, X.P. Comprehensive insights into host-pathogen interaction between brown planthopper and a fungal entomopathogen by dual RNA sequencing. Pest Manag. Sci. 2021, 77, 4903–4914. [Google Scholar] [CrossRef]
- Schmidt, K.; Engel, P. Mechanisms underlying gut microbiota-host interactions in insects. J. Exp. Biol. 2021, 224, jeb207696. [Google Scholar] [CrossRef]
- Jakubowska, A.K.; Vogel, H.; Herrero, S. Increase in gut microbiota after immune suppression in baculovirus-infected larvae. PLoS Pathog. 2013, 9, e1003379. [Google Scholar] [CrossRef]
- Yuan, C.; Xing, L.; Wang, M.; Hu, Z.; Zou, Z. Microbiota modulates gut immunity and promotes baculovirus infection in Helicoverpa armigera. Insect Sci. 2021, 28, 1766–1779. [Google Scholar] [CrossRef]
- Broderick, N.A.; Robinson, C.J.; McMahon, M.D.; Holt, J.; Handelsman, J.; Raffa, K.F. Contributions of gut bacteria to Bacillus thuringiensis-induced mortality vary across a range of Lepidoptera. BMC Biol. 2009, 7, 11. [Google Scholar] [CrossRef] [Green Version]
- Wei, G.; Lai, Y.; Wang, G.; Chen, H.; Li, F.; Wang, S. Insect pathogenic fungus interacts with the gut microbiota to accelerate mosquito mortality. Proc. Natl. Acad. Sci. USA 2017, 114, 5994–5999. [Google Scholar] [CrossRef] [Green Version]
- Steele, M.I.; Motta, E.V.S.; Gattu, T.; Martinez, D.; Moran, N.A. The gut microbiota protects bees from invasion by a bacterial pathogen. Microbiol. Spectr. 2021, 9, e0039421. [Google Scholar] [CrossRef]
- Zhang, F.; Sun, X.X.; Zhang, X.C.; Zhang, S.; Lu, J.; Xia, Y.M.; Huang, Y.H.; Wang, X.J. The interactions between gut microbiota and entomopathogenic fungi: A potential approach for biological control of Blattella germanica (L.). Pest Manag. Sci. 2018, 74, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Knight, R. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2011, 25, 402–408. [Google Scholar] [CrossRef]
- Tang, Q.Y.; Zhang, C.X. Data processing system (DPS) software with experimental design, statistical analysis and data mining developed for use in entomological research. Insect Sci. 2013, 20, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Liberti, J.; Engel, P. The gut microbiota-brain axis of insects. Curr. Opin. Insect Sci. 2020, 39, 6–13. [Google Scholar] [CrossRef]
- Ramirez, J.L.; Souza-Neto, J.; Torres, C.R.; Rovira, J.; Ortiz, A.; Pascale, J.M.; Dimopoulos, G. Reciprocal tripartite interactions between the Aedes aegypti midgut microbiota, innate immune system and dengue virus influences vector competence. PLoS Negl. Trop. Dis. 2012, 6, e1561. [Google Scholar] [CrossRef]
- Yun, J.H.; Roh, S.W.; Whon, T.W.; Jung, M.J.; Kim, M.S.; Park, D.S.; Yoon, C.; Nam, Y.D.; Kim, Y.J.; Choi, J.H.; et al. Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Appl. Environ. Microbiol. 2014, 80, 5254–5264. [Google Scholar] [CrossRef] [Green Version]
- Vijayakumar, M.M.; More, R.P.; Rangasamy, A.; Gandhi, G.R.; Muthugounder, M.; Thiruvengadam, V.; Samaddar, S.; Jalali, S.K.; Sa, T. Gut Bacterial diversity of insecticide-susceptible and -resistant nymphs of the brown planthopper Nilaparvata lugens Stål (Hemiptera: Delphacidae) and elucidation of their putative functional roles. J. Microbiol. Biotechnol. 2018, 28, 976–986. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Tang, T.; Li, W.; Cai, T.; Li, J.; Wan, H. Functional profiling of the gut microbiomes in two different populations of the brown planthopper, Nilaparvata lugens. J. Asia-Pac. Entomol. 2018, 21, 1309–1314. [Google Scholar] [CrossRef]
- Wang, Z.L.; Pan, H.B.; Wu, W.; Li, M.Y.; Yu, X.P. The gut bacterial flora associated with brown planthopper is affected by host rice varieties. Arch. Microbiol. 2021, 203, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Briones-Roblero, C.I.; Rodríguez-Díaz, R.; Santiago-Cruz, J.A.; Zúñiga, G.; Rivera-Orduña, F.N. Degradation capacities of bacteria and yeasts isolated from the gut of Dendroctonus rhizophagus (Curculionidae: Scolytinae). Folia Microbiol. 2017, 62, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Eski, A.; Demir, İ.; Güllü, M.; Demirbağ, Z. Biodiversity and pathogenicity of bacteria associated with the gut microbiota of beet armyworm, Spodoptera exigua Hübner (Lepidoptera: Noctuidae). Microb. Pathog. 2018, 121, 350–358. [Google Scholar] [CrossRef]
- Xu, L.; Deng, J.; Zhou, F.; Cheng, C.; Lu, M. Gut microbiota in an invasive bark beetle infected by a pathogenic fungus accelerates beetle mortality. J. Pest Sci. 2019, 92, 343–351. [Google Scholar] [CrossRef]
- You, H.; Lee, W.J.; Lee, W.J. Homeostasis between gut-associated microorganisms and the immune system in Drosophila. Curr. Opin. Immunol. 2014, 30, 48–53. [Google Scholar] [CrossRef]
- Pang, X.; Xiao, X.; Liu, Y.; Zhang, R.; Liu, J.; Liu, Q.; Wang, P.; Cheng, G. Mosquito C-type lectins maintain gut microbiome homeostasis. Nat. Microbiol. 2016, 1, 16023. [Google Scholar] [CrossRef] [Green Version]
- Bai, S.; Yao, Z.; Raza, M.F.; Cai, Z.; Zhang, H. Regulatory mechanisms of microbial homeostasis in insect gut. Insect Sci. 2021, 28, 286–301. [Google Scholar] [CrossRef]
- Xiao, R.; Wang, X.; Xie, E.; Ji, T.; Li, X.; Muhammad, A.; Yin, X.; Hou, Y.; Shi, Z. An IMD-like pathway mediates the intestinal immunity to modulate the homeostasis of gut microbiota in Rhynchophorus ferrugineus Olivier (Coleoptera: Dryophthoridae). Dev. Comp. Immunol. 2019, 97, 20–27. [Google Scholar] [CrossRef]
- Jang, S.; Mergaert, P.; Ohbayashi, T.; Ishigami, K.; Shigenobu, S.; Itoh, H.; Kikuchi, Y. Dual oxidase enables insect gut symbiosis by mediating respiratory network formation. Proc. Natl. Acad. Sci. USA 2021, 118, e2020922118. [Google Scholar] [CrossRef]
- Ha, E.M.; Lee, K.A.; Seo, Y.Y.; Kim, S.H.; Lim, J.H.; Oh, B.H.; Kim, J.; Lee, W.J. Coordination of multiple dual oxidase-regulatory pathways in responses to commensal and infectious microbes in Drosophila gut. Nat. Immunol. 2009, 10, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Kryukov, V.Y.; Rotskaya, U.; Yaroslavtseva, O.; Polenogova, O.; Kryukova, N.; Akhanaev, Y.; Krivopalov, A.; Alikina, T.; Vorontsova, Y.L.; Slepneva, I.; et al. Fungus Metarhizium robertsii and neurotoxic insecticide affect gut immunity and microbiota in Colorado potato beetles. Sci. Rep. 2021, 11, 1299. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.L.; Dunlap, C.A.; Muturi, E.J.; Barletta, A.B.F.; Rooney, A.P. Entomopathogenic fungal infection leads to temporospatial modulation of the mosquito immune system. PLoS Negl. Trop. Dis. 2018, 12, e0006433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Li, M.; Mao, T.; Wang, H.; Chen, J.; Lu, Z.; Qu, J.; Fang, Y.; Gu, Z.; Li, B. Effects of phoxim exposure on gut microbial composition in the silkworm, Bombyx mori. Ecotoxicol. Environ. Saf. 2020, 189, 110011. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.; Meng, J.; Merchant, A.; Zhang, Y.X.; Li, M.W.; Zhou, X.G.; Wang, Q. Microbial response to fungal infection in a fungus-growing termite, Odontotermes formosanus (Shiraki). Front. Microbiol. 2021, 12, 723508. [Google Scholar] [CrossRef]
- Ríos-Moreno, A.; Garrido-Jurado, I.; Resquín-Romero, G.; Arroyo-Manzanares, N.; Arce, L.; Quesada-Moraga, E. Destruxin A production by Metarhizium brunneum strains during transient endophytic colonisation of Solanum tuberosum. Biocontrol Sci. Technol. 2016, 26, 1574–1585. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Tags | Valid Tags | OTUs | Shannon † | Simpson † | Chao1 † | Coverage † |
|---|---|---|---|---|---|---|---|
| CK | 219,128 | 187,059 | 192 | 3.03 (±0.32) a | 0.76 (±0.01) a | 76.67 (±20.54) a | 0.999 (±0.001) |
| Ma456 | 275,460 | 258,789 | 169 | 1.91 (±0.41) b | 0.59 (±0.06) b | 77.11 (±7.52) a | 1.000 (±0.000) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Cheng, Y.; Wang, Y.; Yu, X. Topical Fungal Infection Induces Shifts in the Gut Microbiota Structure of Brown Planthopper, Nilaparvata lugens (Homoptera: Delphacidae). Insects 2022, 13, 528. https://doi.org/10.3390/insects13060528
Wang Z, Cheng Y, Wang Y, Yu X. Topical Fungal Infection Induces Shifts in the Gut Microbiota Structure of Brown Planthopper, Nilaparvata lugens (Homoptera: Delphacidae). Insects. 2022; 13(6):528. https://doi.org/10.3390/insects13060528
Chicago/Turabian StyleWang, Zhengliang, Yiqing Cheng, Yandan Wang, and Xiaoping Yu. 2022. "Topical Fungal Infection Induces Shifts in the Gut Microbiota Structure of Brown Planthopper, Nilaparvata lugens (Homoptera: Delphacidae)" Insects 13, no. 6: 528. https://doi.org/10.3390/insects13060528