
The Carniolan Honeybee from Slovenia—A Complete and Annotated Mitochondrial Genome with Comparisons to Closely Related Apis mellifera Subspecies

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction
2.3. Whole Genome Sequencing and Assembly
2.4. Complete Mitochondrial Genome Retrieval and Annotation
2.5. Comparisons between Mitochondrial Genomes
2.6. Phylogenetic Analyses
3. Results and Discussion
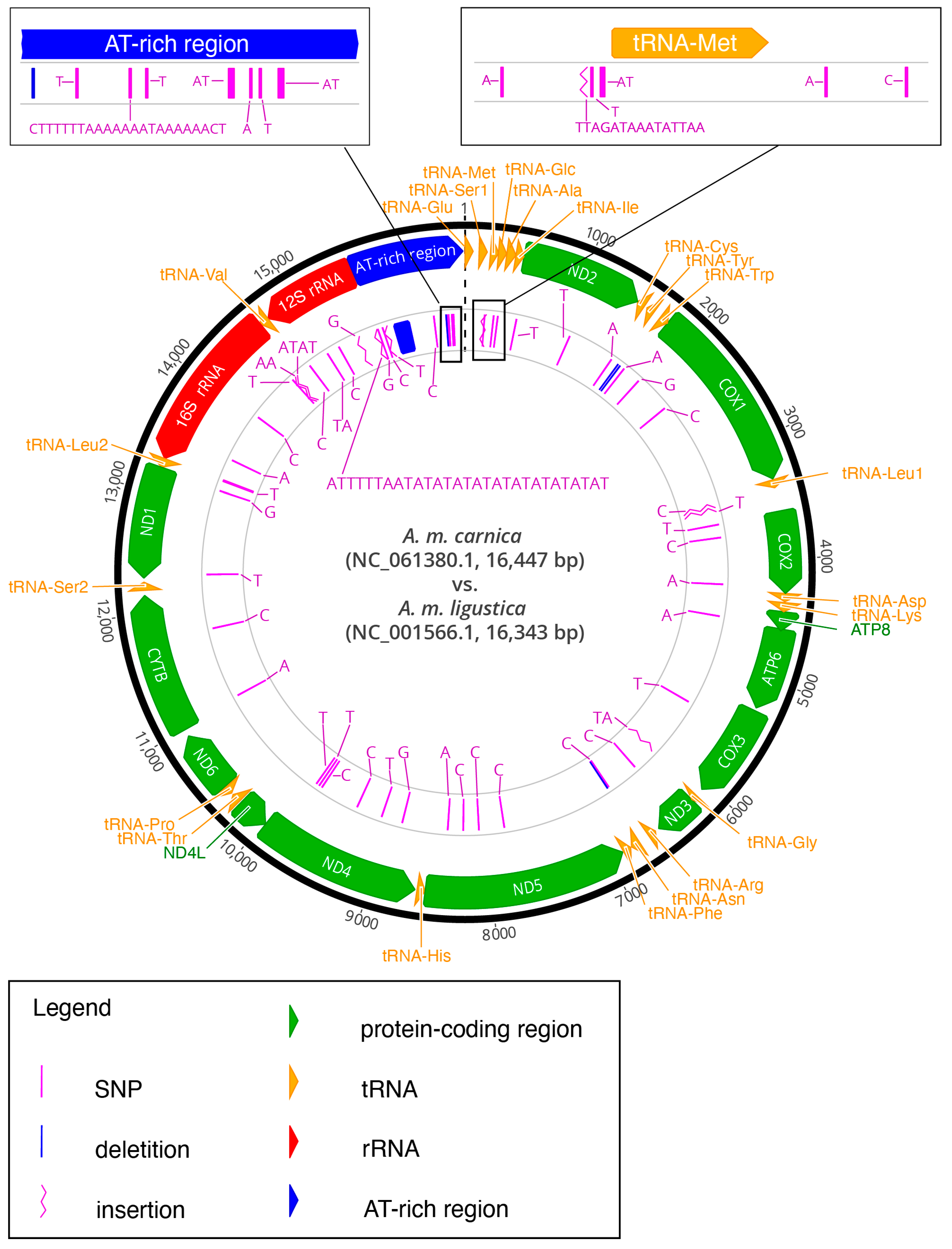
3.1. Main Characteristics of the New Mitochondrial Genome of Apis m. carnica from Slovenia
3.2. Comparison to Mitogenomes from Closely Related Specimens
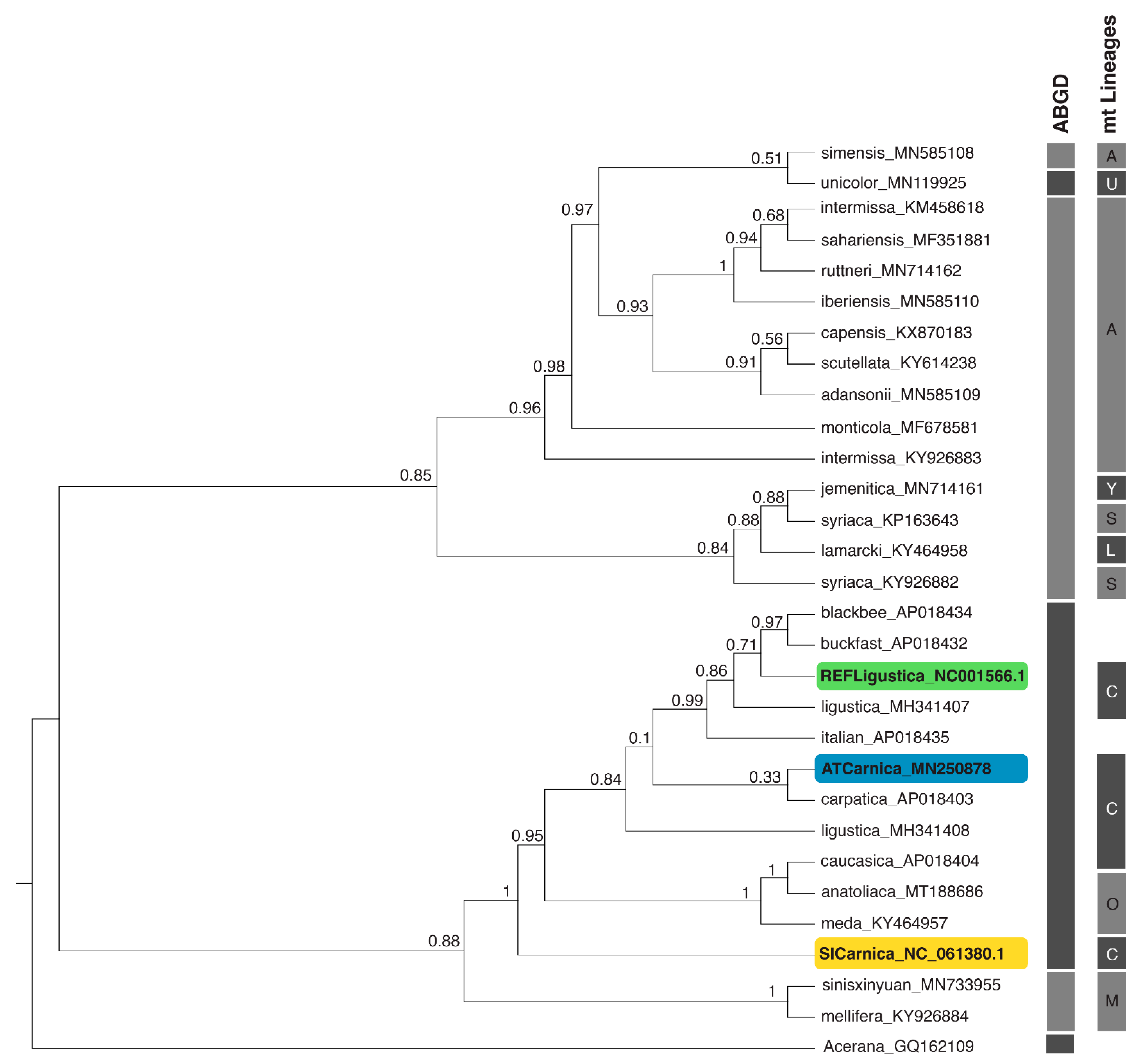
3.3. Phylogenetic Relationships between and within the Evolutionary Lineages
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ruttner, F. Biogeography and Taxonomy of Honeybees; Springer: Berlin/Heidelberg, Germany, 1988. [Google Scholar]
- Sheppard, W.S.; Meixner, M.D. Apis mellifera pomonella, a New Honey Bee Subspecies from Central Asia. Apidologie 2003, 34, 367–375. [Google Scholar] [CrossRef] [Green Version]
- Meixner, M.D.; Leta, M.A.; Koeniger, N.; Fuchs, S. The Honey Bees of Ethiopia Represent a New Subspecies of Apis mellifera—Apis mellifera simensis n. ssp. Apidologie 2011, 42, 425–437. [Google Scholar] [CrossRef]
- Chen, C.; Liu, Z.; Pan, Q.; Chen, X.; Wang, H.; Guo, H.; Liu, S.; Lu, H.; Tian, S.; Li, R.; et al. Genomic Analyses Reveal Demographic History and Temperate Adaptation of the Newly Discovered Honey Bee Subspecies Apis mellifera sinisxinyuan n. ssp. Mol. Biol. Evol. 2016, 33, 1337–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallai, N.; Salles, J.-M.; Settele, J.; Vaissière, B.E. Economic Valuation of the Vulnerability of World Agriculture Confronted with Pollinator Decline. Ecol. Econ. 2009, 68, 810–821. [Google Scholar] [CrossRef]
- Franck, P.; Garnery, L.; Solignac, M.; Cornuet, J.-M. Molecular Confirmation of a Fourth Lineage in Honeybees from the Near East. Apidologie 2000, 31, 167–180. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, C.W.; Behura, S.K.; Berlocher, S.H.; Clark, A.G.; Johnston, J.S.; Sheppard, W.S.; Smith, D.R.; Suarez, A.V.; Weaver, D.; Tsutsui, N.D. Thrice Out of Africa: Ancient and Recent Expansions of the Honey Bee, Apis mellifera. Science 2006, 314, 642–645. [Google Scholar] [CrossRef] [Green Version]
- Alburaki, M.; Bertrand, B.; Legout, H.; Moulin, S.; Alburaki, A.; Sheppard, W.; Garnery, L. A Fifth Major Genetic Group among Honeybees Revealed in Syria. BMC Genet. 2013, 14, 117. [Google Scholar] [CrossRef] [Green Version]
- Tihelka, E.; Cai, C.; Pisani, D.; Donoghue, P.C.J. Mitochondrial Genomes Illuminate the Evolutionary History of the Western Honey Bee (Apis mellifera). Sci. Rep. 2020, 10, 14515. [Google Scholar] [CrossRef]
- Dogantzis, K.A.; Tiwari, T.; Conflitti, I.M.; Dey, A.; Patch, H.M.; Muli, E.M.; Garnery, L.; Whitfield, C.W.; Stolle, E.; Alqarni, A.S.; et al. Thrice out of Asia and the Adaptive Radiation of the Western Honey Bee. Sci. Adv. 2021, 7, eabj2151. [Google Scholar] [CrossRef]
- Garnery, L.; Cornuet, J.-M.; Solignac, M. Evolutionary History of the Honey Bee Apis mellifera Inferred from Mitochondrial DNA Analysis. Mol. Ecol. 1992, 1, 145–154. [Google Scholar] [CrossRef]
- Sušnik, S.; Kozmus, P.; Poklukar, J.; Meglič, V. Molecular Characterisation of Indigenous Apis mellifera carnica in Slovenia. Apidologie 2004, 35, 623–636. [Google Scholar] [CrossRef] [Green Version]
- Ilyasov, R.A.; Lee, M.; Takahashi, J.; Kwon, H.W.; Nikolenko, A.G. A Revision of Subspecies Structure of Western Honey Bee Apis mellifera. Saudi J. Biol. Sci. 2020, 27, 3615–3621. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.G. Animal Mitochondrial DNA as a Genetic Marker in Population and Evolutionary Biology. Trends Ecol. Evol. 1989, 4, 6–11. [Google Scholar] [CrossRef]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA Primers for Amplification of Mitochondrial Cytochrome c Oxidase Subunit I from Diverse Metazoan Invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar]
- Ratnasingham, S.; Hebert, P.D.N. The Barcode of Life Data System. Mol. Ecol. Notes 2007, 7, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Meixner, M.D.; Pinto, M.A.; Bouga, M.; Kryger, P.; Ivanova, E.; Fuchs, S. Standard Methods for Characterising Subspecies and Ecotypes of Apis mellifera. J. Apic. Res. 2013, 52, 1–28. [Google Scholar] [CrossRef]
- Garnery, L.; Solignac, M.; Celebrano, G.; Cornuet, J.-M. A Simple Test Using Restricted PCR-Amplified Mitochondrial DNA to Study the Genetic Structure of Apis mellifera L. Experientia 1993, 49, 1016–1021. [Google Scholar] [CrossRef]
- Henriques, D.; Chavez-Galarza, J.; Quaresma, A.; Neves, C.J.; Lopes, A.R.; Costa, C.; Costa, F.O.; Rufino, J.; Pinto, M.A. From the Popular TRNA(Leu)-COX2 Intergenic Region to the Mitogenome: Insights from Diverse Honey Bee Populations of Europe and North Africa. Apidologie 2019, 50, 215–229. [Google Scholar] [CrossRef] [Green Version]
- Meusel, M.S.; Moritz, R.F.A. Transfer of Paternal Mitochondrial DNA during Fertilization of Honeybee (Apis mellifera L.) Eggs. Curr. Genet. 1993, 24, 539–543. [Google Scholar] [CrossRef]
- Wilson, A.C.; Cann, R.L.; Carr, S.M.; George, M.; Gyllensten, U.B.; Helm-Bychowski, K.M.; Higuchi, R.G.; Palumbi, S.R.; Prager, E.M.; Sage, R.D.; et al. Mitochondrial DNA and Two Perspectives on Evolutionary Genetics. Biol. J. Linn. Soc. 1985, 26, 375–400. [Google Scholar] [CrossRef]
- Cameron, S.L. Insect Mitochondrial Genomics: Implications for Evolution and Phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crozier, R.H.; Crozier, Y.C. The Mitochondrial Genome of the Honeybee Apis mellifera: Complete Sequence and Genome Organization. Genetics 1993, 133, 97–117. [Google Scholar] [CrossRef] [PubMed]
- Ilyasov, R.; Nikolenko, A.; Tuktarov, V.; Goto, K.; Takahashi, J.-I.; Kwon, H.W. Comparative Analysis of Mitochondrial Genomes of the Honey Bee Subspecies A. m. caucasica and A. m. carpathica and Refinement of Their Evolutionary Lineages. J. Apic. Res. 2019, 58, 567–579. [Google Scholar] [CrossRef]
- Okuyama, H.; Hill, J.; Martin, S.J.; Takahashi, J. The Complete Mitochondrial Genome of a Buckfast Bee, Apis mellifera (Insecta: Hymenoptera: Apidae) in Northern Ireland. Mitochondrial DNA Part B 2018, 3, 338–339. [Google Scholar] [CrossRef] [Green Version]
- Maeda, M.; Nakagawa, I.; Chikano, M.; Okuyama, H.; Murray, R.; Takahashi, J.-I. The Complete Mitochondrial Genome of the Dusky Brown-Gray–Colored Honeybee, Apis mellifera (Insecta: Hymenoptera: Apidae) of New Zealand. Mitochondrial DNA Part B 2018, 3, 996–997. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, I.; Maeda, M.; Chikano, M.; Okuyama, H.; Murray, R.; Takahashi, J.-I. The Complete Mitochondrial Genome of the Yellow Coloured Honeybee Apis mellifera (Insecta: Hymenoptera: Apidae) of New Zealand. Mitochondrial DNA Part B 2018, 3, 66–67. [Google Scholar] [CrossRef] [Green Version]
- Hu, P.; Lu, Z.-X.; Haddad, N.; Noureddine, A.; Loucif-Ayad, W.; Wang, Y.-Z.; Zhao, R.-B.; Zhang, A.-L.; Guan, X.; Zhang, H.-X.; et al. Complete Mitochondrial Genome of the Algerian Honeybee, Apis mellifera intermissa (Hymenoptera: Apidae). Mitochondrial DNA Part A 2014, 1–2. [Google Scholar] [CrossRef]
- Haddad, N.J. Mitochondrial Genome of the Levant Region Honeybee, Apis mellifera syriaca (Hymenoptera: Apidae). Mitochondrial DNA Part A 2016, 27, 4067–4068. [Google Scholar] [CrossRef]
- Eimanifar, A.; Kimball, R.T.; Braun, E.L.; Ellis, J.D. The Complete Mitochondrial Genome of the Cape Honey Bee, Apis mellifera capensis esch. (Insecta: Hymenoptera: Apidae). Mitochondrial DNA Part B 2016, 1, 817–819. [Google Scholar] [CrossRef] [Green Version]
- Eimanifar, A.; Kimball, R.T.; Braun, E.L.; Fuchs, S.; Grünewald, B.; Ellis, J.D. The Complete Mitochondrial Genome of Apis mellifera meda (Insecta: Hymenoptera: Apidae). Mitochondrial DNA Part B 2017, 2, 268–269. [Google Scholar] [CrossRef] [Green Version]
- Eimanifar, A.; Kimball, R.T.; Braun, E.L.; Moustafa, D.M.; Haddad, N.; Fuchs, S.; Grünewald, B.; Ellis, J.D. The Complete Mitochondrial Genome of the Egyptian Honey Bee, Apis mellifera lamarckii (Insecta: Hymenoptera: Apidae). Mitochondrial DNA Part B 2017, 2, 270–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haddad, N.; Adjlane, N.; Loucif-Ayad, W.; Dash, A.; Naganeeswaran, S.; Rajashekar, B.; Al-Nakeeb, K.; Sicheritz-Ponten, T. Mitochondrial Genome of the North African Sahara Honeybee, Apis mellifera sahariensis (Hymenoptera: Apidae). Mitochondrial DNA Part B 2017, 2, 548–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eimanifar, A.; Kimball, R.T.; Braun, E.L.; Fuchs, S.; Grünewald, B.; Ellis, J.D. The Complete Mitochondrial Genome of an East African Honey Bee, Apis mellifera monticola Smith (Insecta: Hymenoptera: Apidae). Mitochondrial DNA Part B 2017, 2, 589–590. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; Wang, A.R.; Kim, M.J.; Lee, K.H.; Kim, I. Single-Nucleotide Polymorphism Markers in Mitochondrial Genomes for Identifying Varroa destructor -Resistant and -Susceptible Strains of Apis mellifera (Hymenoptera: Apidae). Mitochondrial DNA Part A 2019, 30, 477–489. [Google Scholar] [CrossRef]
- Boardman, L.; Eimanifar, A.; Kimball, R.T.; Braun, E.L.; Fuchs, S.; Grünewald, B.; Ellis, J.D. The Complete Mitochondrial Genome of Apis mellifera unicolor (Insecta: Hymenoptera: Apidae), the Malagasy Honey Bee. Mitochondrial DNA Part B 2019, 4, 3286–3287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boardman, L.; Eimanifar, A.; Kimball, R.T.; Braun, E.L.; Fuchs, S.; Grünewald, B.; Ellis, J.D. The Mitochondrial Genome of the Carniolan Honey Bee, Apis mellifera carnica (Insecta: Hymenoptera: Apidae). Mitochondrial DNA Part B 2019, 4, 3288–3290. [Google Scholar] [CrossRef] [Green Version]
- Boardman, L.; Eimanifar, A.; Kimball, R.; Braun, E.; Fuchs, S.; Grünewald, B.; Ellis, J.D. The Mitochondrial Genome of Apis mellifera simensis (Hymenoptera: Apidae), an Ethiopian Honey Bee. Mitochondrial DNA Part B 2020, 5, 9–10. [Google Scholar] [CrossRef] [Green Version]
- Boardman, L.; Eimanifar, A.; Kimball, R.; Braun, E.; Fuchs, S.; Grünewald, B.; Ellis, J.D. The Complete Mitochondrial Genome of the West African Honey Bee Apis mellifera adansonii (Insecta: Hymenoptera: Apidae). Mitochondrial DNA Part B 2020, 5, 11–12. [Google Scholar] [CrossRef] [Green Version]
- Boardman, L.; Eimanifar, A.; Kimball, R.; Braun, E.; Fuchs, S.; Grünewald, B.; Ellis, J.D. The Mitochondrial Genome of the Spanish Honey Bee, Apis mellifera iberiensis (Insecta: Hymenoptera: Apidae), from Portugal. Mitochondrial DNA Part B 2020, 5, 17–18. [Google Scholar] [CrossRef] [Green Version]
- Boardman, L.; Eimanifar, A.; Kimball, R.T.; Braun, E.L.; Fuchs, S.; Grünewald, B.; Ellis, J.D. The Complete Mitochondrial Genome of Apis mellifera jemenitica (Insecta: Hymenoptera: Apidae), the Arabian Honey Bee. Mitochondrial DNA Part B 2020, 5, 875–876. [Google Scholar] [CrossRef] [Green Version]
- Boardman, L.; Eimanifar, A.; Kimball, R.T.; Braun, E.L.; Fuchs, S.; Grünewald, B.; Ellis, J.D. The Mitochondrial Genome of the Maltese Honey Bee, Apis mellifera ruttneri (Insecta: Hymenoptera: Apidae). Mitochondrial DNA Part B 2020, 5, 877–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Jiao, Z.; Wen, X.; Liu, B.; Huang, J.; Ding, G. The Complete Mitochondrial Genome of the Xinyuan Honey Bee, Apis mellifera sinisxinyuan (Insecta: Hymenoptera: Apidae). Mitochondrial DNA Part B 2020, 5, 486–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boardman, L.; Kimball, R.T.; Braun, E.L.; Fuchs, S.; Grünewald, B.; Ellis, J.D. Mitochondrial Genome of Apis mellifera anatoliaca (Hymenoptera: Apidae)–the Anatolian Honey Bee. Mitochondrial DNA Part B 2020, 5, 1876–1877. [Google Scholar] [CrossRef] [Green Version]
- Eimanifar, A.; Kimball, R.T.; Braun, E.L.; Ellis, J.D. Mitochondrial Genome Diversity and Population Structure of Two Western Honey Bee Subspecies in the Republic of South Africa. Sci. Rep. 2018, 8, 1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuller, Z.L.; Niño, E.L.; Patch, H.M.; Bedoya-Reina, O.C.; Baumgarten, T.; Muli, E.; Mumoki, F.; Ratan, A.; McGraw, J.; Frazier, M.; et al. Genome-Wide Analysis of Signatures of Selection in Populations of African Honey Bees (Apis mellifera) Using New Web-Based Tools. BMC Genom. 2015, 16, 518. [Google Scholar] [CrossRef] [Green Version]
- Commission on Genetic Resources for Food and Agriculture. Global Survey of Honeybees and Other Pollinators. 2018. Available online: https://www.cbd.int/doc/c/e5b4/ac7d/15f592d0af6224900c95d04b/sbstta-22-inf-16-en.pdf (accessed on 1 April 2022).
- Nazzi, F. Morphometric Analysis of Honey Bees from an Area of Racial Hybridization in Northeastern Italy. Apidologie 1992, 23, 89–96. [Google Scholar] [CrossRef] [Green Version]
- Péntek-Zakar, E.; Oleksa, A.; Borowik, T.; Kusza, S. Population Structure of Honey Bees in the Carpathian Basin (Hungary) Confirms Introgression from Surrounding Subspecies. Ecol. Evol. 2015, 5, 5456–5467. [Google Scholar] [CrossRef] [Green Version]
- Dedej, S.; Biasiolo, A.; Piva, R. Morphometric and Alloenzymatic Characterisation in the Albanian Honeybee Population Apis mellifera L. Apidologie 1996, 27, 121–131. [Google Scholar] [CrossRef] [Green Version]
- Mihelič, S. Der Mensch Und Die Biene; Der Slowenischen Ethnographischen Museums Und Des Osterreichischen Museums Fur Volkskunde: Ljubljana, Slovenia; Wien, Austria, 1989. [Google Scholar]
- European Comission. Final Act to the Treaty of Accession to the European Union 2003, Declaration by the Republic of Slovenia on the Slovenian Indigenous Bee Apis mellifera carnica (Kranjska Čebela). Official Journal of the European Union, L236, 984. 2003. Available online: https://www.fdfa.be/sites/default/files/atoms/files/564_Final%20act%20in%20English.pdf (accessed on 1 April 2022).
- Kozmus, P.; Podgoršek, P.; Smodiš Škerl, M.I.; Prešern, J.; Metelko, M.; Hrastelj, M. Rejski Program za Kranjsko Čebelo (Apis mellifera carnica); Agricultural Institute of Slovenia: Ljubljana, Slovenia, 2018. [Google Scholar]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of Long, Error-Prone Reads Using Repeat Graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and Accurate de Novo Genome Assembly from Long Uncorrected Reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Medaka; Oxford Nanopore Technologies: Oxford, UK, 2021.
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Jackman, S.D.; Coombe, L.; Chu, J.; Warren, R.L.; Vandervalk, B.P.; Yeo, S.; Xue, Z.; Mohamadi, H.; Bohlmann, J.; Jones, S.J.M.; et al. Tigmint: Correcting Assembly Errors Using Linked Reads from Large Molecules. BMC Bioinform. 2018, 19, 393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness. In Gene Prediction; Kollmar, M., Ed.; Springer: New York, NY, USA, 2019; Volume 1962, pp. 227–245. [Google Scholar]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Shumate, A.; Salzberg, S.L. Liftoff: Accurate Mapping of Gene Annotations. Bioinformatics 2021, 37, 1639–1643. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laslett, D.; Canback, B. ARWEN: A Program to Detect TRNA Genes in Metazoan Mitochondrial Nucleotide Sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-Specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.-W.; Liu, G.-H.; Dong, X.; Lin, R.-Q.; Song, H.-Q.; Huang, S.-Y.; Yuan, Z.-G.; Zhao, G.-H.; Zhu, X.-Q. The Complete Mitochondrial Genome of the Asiatic Cavity-Nesting Honeybee Apis Cerana (Hymenoptera: Apidae). PLoS ONE 2011, 6, e23008. [Google Scholar] [CrossRef] [Green Version]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [Green Version]
- Dress, A.W.; Flamm, C.; Fritzsch, G.; Grünewald, S.; Kruspe, M.; Prohaska, S.J.; Stadler, P.F. Noisy: Identification of Problematic Columns in Multiple Sequence Alignments. Algorithms Mol. Biol. 2008, 3, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for Inference of Large Phylogenetic Trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A Fast, Scalable and User-Friendly Tool for Maximum Likelihood Phylogenetic Inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lartillot, N.; Lepage, T.; Blanquart, S. PhyloBayes 3: A Bayesian Software Package for Phylogenetic Reconstruction and Molecular Dating. Bioinformatics 2009, 25, 2286–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. JModelTest 2: More Models, New Heuristics and Parallel Computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Gascuel, O. A Simple, Fast, and Accurate Algorithm to Estimate Large Phylogenies by Maximum Likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Jukes, T.H.; Cantor, C.R. Evolution of Protein Molecules. In Mammalian Protein Metabolism; Munro, H.N., Ed.; Academic Press: Cambridge, MA, USA, 1969; Volume 3. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Puillandre, N.; Lambert, A.; Brouillet, S.; Achaz, G. ABGD, Automatic Barcode Gap Discovery for Primary Species Delimitation: ABGD, Automatic Barcode Gap Discovery. Mol. Ecol. 2012, 21, 1864–1877. [Google Scholar] [CrossRef]
- Kimura, M. A Simple Method for Estimating Evolutionary Rates of Base Substitutions through Comparative Studies of Nucleotide Sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Firestone, K.B. Phylogenetic Relationships Among Quolls Revisited: The MtDNA Control Region as a Useful Tool. J. Mamm. Evol. 2000, 7, 1–22. [Google Scholar] [CrossRef]
- Kozmus, P.; Stevanović, J.; Stanimirović, Z.; Stojić, V.; Kulišić, Z.; Meglič, V. Analysis of Mitochondrial DNA in Honey Bees (Apis mellifera) from Serbia. Acta Vet. 2007, 57, 465–476. [Google Scholar] [CrossRef]
- Muñoz, I.; Stevanovic, J.; Stanimirovic, Z.; De la Rúa, P. Genetic Variation of from Serbia Inferred from Mitochondrial Analysis. J. Apic. Sci. 2012, 56, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.H.; Tu, F.Y. Characterization and Evolution of the Mitochondrial DNA Control Region in Ranidae and Their Phylogenetic Relationship. Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef] [PubMed]
- Bronstein, O.; Kroh, A.; Haring, E. Mind the Gap! The Mitochondrial Control Region and Its Power as a Phylogenetic Marker in Echinoids. BMC Evol. Biol. 2018, 18, 80. [Google Scholar] [CrossRef] [PubMed]
- Diniz, F.M.; Maclean, N.; Ogawa, M.; Cintra, I.H.A.; Bentzen, P. The Hypervariable Domain of the Mitochondrial Control Region in Atlantic Spiny Lobsters and Its Potential as a Marker for Investigating Phylogeographic Structuring. Mar. Biotechnol. 2005, 7, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Fenn, J.D.; Cameron, S.L.; Whiting, M.F. The Complete Mitochondrial Genome Sequence of the Mormon Cricket (Anabrus Simplex: Tettigoniidae: Orthoptera) and an Analysis of Control Region Variability. Insect Mol. Biol. 2007, 16, 239–252. [Google Scholar] [CrossRef]
- Zhang, D.-X.; Hewitt, G.M. Insect Mitochondrial Control Region: A Review of Its Structure, Evolution and Usefulness in Evolutionary Studies. Biochem. Syst. Ecol. 1997, 25, 99–120. [Google Scholar] [CrossRef]
- Gonçalves, R.; Freitas, A.I.; Jesus, J.; De la Rúa, P.; Brehm, A. Structure and Genetic Variation of the Mitochondrial Control Region in the Honey Bee Apis mellifera. Apidologie 2015, 46, 515–526. [Google Scholar] [CrossRef] [Green Version]
- Zorc, M.; Božič, J.; Dovč, P. Genome Sequence Variation in Two Subspecies of Western Honeybee, A.m.Carnica and A.m.Ligustica. Acta Fytotechn Zootech. 2020, 23, 331–337. [Google Scholar] [CrossRef]
- Wagner, G.P. A Comparative Study of Morphological Integration in Apis mellifera (Insecta, Hymenoptera). J. Zool. Syst. Evol. Res. 1990, 28, 48–61. [Google Scholar] [CrossRef]
- Al-Ghamdi, A.A.; Nuru, A.; Khanbash, M.S.; Smith, D.R. Geographical Distribution and Population Variation of Apis mellifera Jemenitica Ruttner. J. Apic. Res. 2013, 52, 124–133. [Google Scholar] [CrossRef] [Green Version]
- Kandemir, I.; Kence, M.; Kence, A. Genetic and Morphometric Variation in Honeybee ( Apis mellifera L.) Populations of Turkey. Apidologie 2000, 31, 343–356. [Google Scholar] [CrossRef] [Green Version]
- Kandemir, I.; Kence, M.; Sheppard, W.S.; Kence, A. Mitochondrial DNA Variation in Honey Bee (Apis mellifera L.) Populations from Turkey. J. Apic. Res. 2006, 45, 33–38. [Google Scholar] [CrossRef]
- Kandemir, İ.; Özkan, A.; Fuchs, S. Reevaluation of Honeybee (Apis mellifera) Microtaxonomy: A Geometric Morphometric Approach. Apidologie 2011, 42, 618–627. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Hu, R.; Costa, C.; Li, J. Genetic Drift and Purifying Selection Shaped Mitochondrial Genome Variation in the High Royal Jelly-Producing Honeybee Strain (Apis mellifera Ligustica). Front. Genet. 2022, 13, 835967. [Google Scholar] [CrossRef]
- Sutarno; Cummins, J.M.; Greeff, J.; Lymbery, A.J. Mitochondrial DNA Polymorphisms and Fertility in Beef Cattle. Theriogenology 2002, 57, 1603–1610. [Google Scholar] [CrossRef]
- Mannen, H.; Morimoto, M.; Oyama, K.; Mukai, F.; Tsuji, S. Identification of Mitochondrial DNA Substitutions Related to Meat Quality in Japanese Black Cattle 1,2. J. Anim. Sci. 2003, 81, 68–73. [Google Scholar] [CrossRef]
- Moškrič, A.; Formato, G.; Škerl, M.I.S.; Prešern, J. Reproductive Technologies in the Honeybee (Apis mellifera). In Reproductive Technologies in Animals; Elsevier: Amsterdam, The Netherlands, 2020; pp. 229–243. [Google Scholar]
- Hedges, C.P.; Wilkinson, R.T.; Devaux, J.B.L.; Hickey, A.J.R. Hymenoptera Flight Muscle Mitochondrial Function: Increasing Metabolic Power Increases Oxidative Stress. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2019, 230, 115–121. [Google Scholar] [CrossRef]
- Gençer, H.V.; Kahya, Y. Sperm Competition in Honey Bees (Apis mellifera L.): The Role of Body Size Dimorphism in Drones. Apidologie 2020, 51, 1–17. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Aln Position (bp) | Region | SICarnica Position (bp) | ATCarnica Position (bp) | REFLigustica Position (bp) | Amino Acid Change | |||
|---|---|---|---|---|---|---|---|---|
| 1113 | nd2 | 1095 | A | 1113 | T | 1110 | T | L → F |
| 1944 | cox1 | 1926 | A | 1936 | G | 1933 | G | N → S |
| 5506 | cox3 | 5486 | A | 5497 | T | 5495 | T | T → S |
| 7849 | nd5 | 7821 | A | 7840 | C | 7830 | C | S → A |
| 9823 | nd4 | 9795 | C | 9814 | T | 9804 | T | D → N |
| 9854 | nd4 | 9826 | A | 9845 | T | 9835 | T | F → L |
| 11,070 | cytb | 11,042 | T | 11,061 | A | 11,051 | A | I → M |
| 12,344 | nd1 | 12,316 | C | 12,335 | T | 12,325 | T | V → M |
| Aln Position (bp) | Region | SICarnica Position (bp) | ATCarnica Position (bp) | REFLigustica Position (bp) | Amino Acid Change (i.a.) | |||
|---|---|---|---|---|---|---|---|---|
| 218 | IR1 | 202 | A | 218 | A | 217 | - | |
| 544 | nd2 | 526 | C | 544 | C | 541 | T | L → F |
| 1592 | IR2 | 1574 | G | 1592 | G | 1589 | A | |
| 3439 | IR3 | 3420 | - | 3430 | - | 3428 | C | |
| 4253 | cox2 | 4233 | G | 4244 | G | 4242 | A | V → I |
| 4577 | atp8 | 4557 | G | 4568 | G | 4566 | A | |
| 6376 | nd3 | 6348 | T | 6367 | T | 6359 | C | |
| 6712 | IR4 | 6684 | AT | 6703 | AT | 6694 | -- | |
| 8108 | nd5 | 8080 | T | 8099 | T | 8089 | C | |
| 8894 | nd4 | 8866 | A | 8885 | A | 8875 | G | |
| 9110 | nd4 | 9082 | A | 9101 | A | 9091 | T | F → L |
| 11,796 | cytb | 11,768 | T | 11,787 | T | 11,777 | C | |
| 13,203 | nd1 | 13,175 | A | 13,194 | A | 13,184 | G | |
| 13,312 | 16S rRNA | 13,284 | C | 13,303 | C | 13,293 | T | |
| 14,046 | 16S rRNA | 14,018 | T | 14,035 | T | 14,025 | C | |
| 14,568 | 16S rRNA | 14,540 | C | 14,557 | C | 14,547 | T | |
| 14,589 | 16S rRNA | 14,559 | - | 14,578 | T | 14,568 | A | |
| 15,150 | 12S rRNA | 15,115 | A | 15,139 | A | 15,129 | C | |
| 15,361 | 12S rRNA | 15,325 | - | 15,349 | - | 15,340 | G | |
| 15,613 | CR | 15,547 | T | 15,601 | T | 15,591 | G | |
| 16,204 | CR | 16,135 | T | 16,020 | T | 16,010 | C | |
| 16,325 | CR | 16,255 | A | 16,141 | A | 16,129 | - | |
| 16,354 | CR | 16,284 | A | 16,170 | A | 16,158 | C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moškrič, A.; Marinč, A.; Ferk, P.; Leskošek, B.; Mosbech, M.-B.; Bunikis, I.; Pettersson, O.V.; Soler, L.; Prešern, J. The Carniolan Honeybee from Slovenia—A Complete and Annotated Mitochondrial Genome with Comparisons to Closely Related Apis mellifera Subspecies. Insects 2022, 13, 403. https://doi.org/10.3390/insects13050403
Moškrič A, Marinč A, Ferk P, Leskošek B, Mosbech M-B, Bunikis I, Pettersson OV, Soler L, Prešern J. The Carniolan Honeybee from Slovenia—A Complete and Annotated Mitochondrial Genome with Comparisons to Closely Related Apis mellifera Subspecies. Insects. 2022; 13(5):403. https://doi.org/10.3390/insects13050403
Chicago/Turabian StyleMoškrič, Ajda, Andraž Marinč, Polonca Ferk, Brane Leskošek, Mai-Britt Mosbech, Ignas Bunikis, Olga Vinnere Pettersson, Lucile Soler, and Janez Prešern. 2022. "The Carniolan Honeybee from Slovenia—A Complete and Annotated Mitochondrial Genome with Comparisons to Closely Related Apis mellifera Subspecies" Insects 13, no. 5: 403. https://doi.org/10.3390/insects13050403