Bacterial Communities in the Feces of Laboratory Reared Gampsocleis gratiosa (Orthoptera: Tettigoniidae) across Different Developmental Stages and Sexes


Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
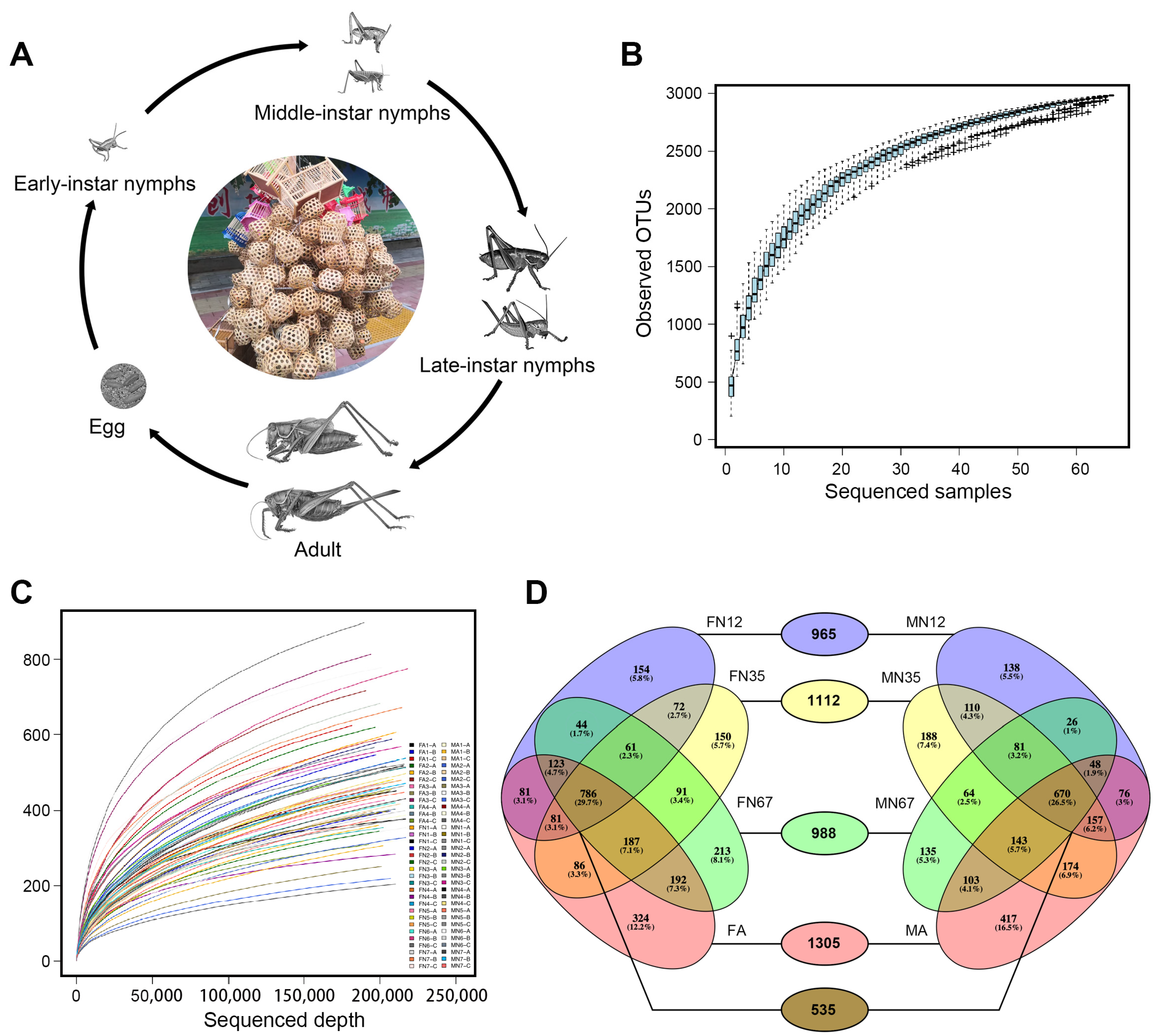
2.1. Insect Rearing and Fecal Sample Collection
2.2. DNA Extraction, 16S rDNA V3–V4 Amplification and Illumina Sequencing
2.3. Bioinformatics and Statistical Analysis
3. Results
3.1. Sequencing Statistics
3.2. Bacterial Community Structures and OTUs
3.3. Dominant and Core Bacterial OTUs
3.4. Alpha and Beta Diversity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Engel, P.; Moran, N.A. The gut microbiota of insects—Diversity in structure and function. FEMS Microbiol. Rev. 2013, 37, 699–735. [Google Scholar] [CrossRef] [PubMed]
- Douglas, A.E. Nutritional interactions in insect-microbial symbioses: Aphids and their symbiotic bacteria Buchnera. Annu. Rev. Entomol. 1998, 43, 17–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, C.M.; Hunter, M.S. Extraordinarily widespread and fantastically complex: Comparative biology of endosymbiotic bacterial and fungal mutualists of insects. Ecol. Lett. 2010, 13, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Tagliavia, M.; Messina, E.; Manachini, B.; Cappello, S.; Quatrini, P. The gut microbiota of larvae of Rhynchophorus ferrugineus Oliver (Coleoptera: Curculionidae). BMC Microbiol. 2014, 14, 136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berlanga, M.; Llorens, C.; Comas, J.; Guerrero, R. Gut bacterial community of the xylophagous cockroaches Cryptocercus punctulatus and Parasphaeria boleiriana. PLoS ONE 2016, 11, e0152400. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Teh, B.S.; Sun, C.; Hu, S.; Lu, X.; Boland, W.; Shao, Y. Biodiversity and activity of the gut microbiota across the life history of the insect herbivore Spodoptera littoralis. Sci. Rep. 2016, 6, 29505. [Google Scholar] [CrossRef]
- Chen, B.; Du, K.; Sun, C.; Vimalanathan, A.; Liang, X.; Li, Y.; Wang, B.; Lu, X.; Li, L.; Shao, Y. Gut bacterial and fungal communities of the domesticated silkworm (Bombyx mori) and wild mulberry-feeding relatives. ISME J. 2018, 12, 2252–2262. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Shu, J.; Xue, H.; Zhang, W.; Zhang, Y.; Liu, Y.; Fang, L.; Wang, Y.; Wang, H. The gut microbiota in camellia weevils are influenced by plant secondary metabolites and contribute to saponin degradation. mSystems 2020, 5, e00692-e19. [Google Scholar] [CrossRef] [Green Version]
- Yun, J.H.; Roh, S.W.; Whon, T.W.; Jung, M.J.; Kim, M.S.; Park, D.S.; Yoon, C.; Nam, Y.D.; Kim, Y.J.; Choi, J.H.; et al. Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Appl. Environ. Microbiol. 2014, 80, 5254–5264. [Google Scholar] [CrossRef] [Green Version]
- Tinker, K.A.; Ottesen, E.A. Phylosymbiosis across deeply diverging lineages in omnivorous cockroaches. Appl. Environ. Microbiol. 2020, 86, e02513-19. [Google Scholar] [CrossRef]
- Schauer, C.; Thompson, C.L.; Brune, A. The bacterial community in the gut of the Cockroach Shelfordella lateralis reflects the close evolutionary relatedness of cockroaches and termites. Appl. Environ. Microbiol. 2012, 78, 2758–2767. [Google Scholar] [CrossRef] [Green Version]
- Perez-Cobas, A.E.; Maiques, E.; Angelova, A.; Carrasco, P.; Moya, A.; Latorre, A. Diet shapes the gut microbiota of the omnivorous cockroach Blattella germanica. FEMS Microbiol. Ecol. 2015, 91, fiv022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertino-Grimaldi, D.; Medeiros, M.N.; Vieira, R.P.; Cardoso, A.M.; Turque, A.S.; Silveira, C.B.; Albano, R.M.; Bressan-Nascimento, S.; Garcia, E.S.; de Souza, W.; et al. Bacterial community composition shifts in the gut of Periplaneta americana fed on different lignocellulosic materials. Springerplus 2013, 2, 609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wu, N.; Liu, Y.; Kundu, J.K.; Liu, W.; Wang, X. Higher bacterial diversity of gut microbiota in different natural populations of leafhopper vector does not influence WDV transmission. Front. Microbiol. 2019, 10, 1144. [Google Scholar] [CrossRef] [Green Version]
- Kuechler, S.M.; Renz, P.; Dettner, K.; Kehl, S. Diversity of symbiotic organs and bacterial endosymbionts of lygaeoid bugs of the families Blissidae and Lygaeidae (Hemiptera: Heteroptera: Lygaeoidea). Appl. Environ. Microbiol. 2012, 78, 2648–2659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaltenpoth, M.; Winter, S.A.; Kleinhammer, A. Localization and transmission route of Coriobacterium glomerans, the endosymbiont of pyrrhocorid bugs. FEMS Microbiol. Ecol. 2009, 69, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Paniagua Voirol, L.R.; Frago, E.; Kaltenpoth, M.; Hilker, M.; Fatouros, N.E. Bacterial symbionts in Lepidoptera: Their diversity, transmission, and impact on the host. Front. Microbiol. 2018, 9, 556. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Hosokawa, T.; Fukatsu, T. Insect-microbe mutualism without vertical transmission: A stinkbug acquires a beneficial gut symbiont from the environment every generation. Appl. Environ. Microbiol. 2007, 73, 4308–4316. [Google Scholar] [CrossRef] [Green Version]
- Hammer, T.J.; Janzen, D.H.; Hallwachs, W.; Jaffe, S.P.; Fierer, N. Caterpillars lack a resident gut microbiome. Proc. Natl. Acad. Sci. USA 2017, 114, 9641–9646. [Google Scholar] [CrossRef] [Green Version]
- Augustinos, A.A.; Tsiamis, G.; Caceres, C.; Abd-Alla, A.M.M.; Bourtzis, K. Taxonomy, diet, and developmental stage contribute to the structuring of gut-associated bacterial communities in Tephritid pest species. Front. Microbiol. 2019, 10, 2004. [Google Scholar] [CrossRef] [Green Version]
- Andongma, A.A.; Wan, L.; Dong, Y.C.; Li, P.; Desneux, N.; White, J.A.; Niu, C.Y. Pyrosequencing reveals a shift in symbiotic bacteria populations across life stages of Bactrocera dorsalis. Sci. Rep. 2015, 5, 9470. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.P.; Sanders, J.G.; Byrne, M.J.; Pierce, N.E. Gut microbiota of dung beetles correspond to dietary specializations of adults and larvae. Mol. Ecol. 2016, 25, 6092–6106. [Google Scholar] [CrossRef] [PubMed]
- Ventura, C.; Briones-Roblero, C.I.; Hernandez, E.; Rivera-Orduna, F.N.; Zuniga, G. Comparative analysis of the gut bacterial community of four Anastrepha fruit flies (Diptera: Tephritidae) based on Pyrosequencing. Curr. Microbiol. 2018, 75, 966–976. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gao, Q.; Wang, W.; Wang, X.; Lei, C.; Zhu, F. The gut bacteria across life stages in the synanthropic fly Chrysomya megacephala. BMC Microbiol. 2018, 18, 131. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.C.; Kim, S.H.; You, H.; Kim, B.; Kim, A.C.; Lee, K.A.; Yoon, J.H.; Ryu, J.H.; Lee, W.J. Drosophila microbiome modulates host developmental and metabolic homeostasis via insulin signaling. Science 2011, 334, 670–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storelli, G.; Defaye, A.; Erkosar, B.; Hols, P.; Royet, J.; Leulier, F. Lactobacillus plantarum promotes Drosophila systemic growth by modulating hormonal signals through TOR-dependent nutrient sensing. Cell Metab. 2011, 14, 403–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, C.M.; Darling, A.E. A method for high precision sequencing of near full-length 16S rRNA genes on an Illumina MiSeq. PeerJ 2016, 4, e2492. [Google Scholar] [CrossRef]
- Gonzalez-Escobedo, R.; Briones-Roblero, C.I.; Pineda-Mendoza, R.M.; Rivera-Orduna, F.N.; Zuniga, G. Bacteriome from Pinus arizonica and P. durangensis: Diversity, comparison of assemblages, and overlapping degree with the gut bacterial community of a bark beetle that kills pines. Front. Microbiol. 2018, 9, 77. [Google Scholar] [CrossRef]
- Zhang, Z.; Jiao, S.; Li, X.; Li, M. Bacterial and fungal gut communities of Agrilus mali at different developmental stages and fed different diets. Sci. Rep. 2018, 8, 15634. [Google Scholar] [CrossRef] [Green Version]
- Rimoldi, S.; Gini, E.; Iannini, F.; Gasco, L.; Terova, G. The effects of dietary insect meal from Hermetia illucens prepupae on autochthonous gut microbiota of rainbow trout (Oncorhynchus mykiss). Animals 2019, 9, 143. [Google Scholar] [CrossRef] [Green Version]
- Dubois, G.; Girard, C.; Lapointe, F.J.; Shapiro, B.J. The Inuit gut microbiome is dynamic over time and shaped by traditional foods. Microbiome 2017, 5, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Wang, W.; Zhu, F.; Wang, X.; Wang, X.; Lei, C. The gut microbiota in larvae of the housefly Musca domestica and their horizontal transfer through feeding. AMB Express 2017, 7, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales-Jimenez, J.; Zuniga, G.; Ramirez-Saad, H.C.; Hernandez-Rodriguez, C. Gut-associated bacteria throughout the life cycle of the bark beetle Dendroctonus rhizophagus Thomas and Bright (Curculionidae: Scolytinae) and their cellulolytic activities. Microb. Ecol. 2012, 64, 268–278. [Google Scholar] [CrossRef]
- Kim, J.M.; Choi, M.Y.; Kim, J.W.; Lee, S.A.; Ahn, J.H.; Song, J.; Kim, S.H.; Weon, H.Y. Effects of diet type, developmental stage, and gut compartment in the gut bacterial communities of two Cerambycidae species (Coleoptera). J. Microbiol. 2017, 55, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Muratore, M.; Prather, C.; Sun, Y. The gut bacterial communities across six grasshopper species from a coastal tallgrass prairie. PLoS ONE 2020, 15, e0228406. [Google Scholar] [CrossRef] [Green Version]
- Muratore, M.; Sun, Y.; Prather, C. Environmental nutrients alter bacterial and fungal gut microbiomes in the common meadow katydid, Orchelimum vulgare. Front. Microbiol. 2020, 11, 557980. [Google Scholar] [CrossRef]
- Zheng, X.; Zhu, Q.; Zhou, Z.; Wu, F.; Chen, L.; Cao, Q.; Shi, F. Gut bacterial communities across 12 Ensifera (Orthoptera) at different feeding habits and its prediction for the insect with contrasting feeding habits. PLoS ONE 2021, 16, e0250675. [Google Scholar] [CrossRef]
- Dillon, R.; Charnley, K. Mutualism between the desert locust Schistocerca gregaria and its gut microbiota. Res. Microbiol. 2002, 153, 503–509. [Google Scholar] [CrossRef]
- Takahashi, S.; Tomita, J.; Nishioka, K.; Hisada, T.; Nishijima, M. Development of a prokaryotic universal primer for simultaneous analysis of Bacteria and Archaea using next-generation sequencing. PLoS ONE 2014, 9, e105592. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef] [Green Version]
- Kruskal, W.H.; Wallis, W.A. Use of ranks in one-criterion variance analysis. J. Am. Stat. Assoc. 1952, 47, 583–621. [Google Scholar] [CrossRef]
- Bray, J.R.; Curtis, J.T. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navas-Molina, J.A.; Peralta-Sanchez, J.M.; Gonzalez, A.; McMurdie, P.J.; Vazquez-Baeza, Y.; Xu, Z.J.; Ursell, L.K.; Lauber, C.; Zhou, H.W.; Song, S.J.; et al. Advancing our understanding of the human microbiome using QIIME. Methods Enzymol. 2013, 531, 371–444. [Google Scholar]
- Jing, T.Z.; Qi, F.H.; Wang, Z.Y. Most dominant roles of insect gut bacteria: Digestion, detoxification, or essential nutrient provision? Microbiome 2020, 8, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.; Nair, S. Dynamics of insect-microbiome interaction influence host and microbial symbiont. Front. Microbiol. 2020, 11, 1357. [Google Scholar] [CrossRef] [PubMed]
- Hegde, S.; Khanipov, K.; Albayrak, L.; Golovko, G.; Pimenova, M.; Saldana, M.A.; Rojas, M.M.; Hornett, E.A.; Motl, G.C.; Fredregill, C.L.; et al. Microbiome interaction networks and community structure from laboratory-reared and field-collected Aedes aegypti, Aedes albopictus, and Culex quinquefasciatus mosquito vectors. Front. Microbiol. 2018, 9, 2160. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Zheng, X.; Long, H.; Rao, Z.; Cao, L.; Han, R. Gut bacterial and fungal communities of the wild and laboratory-reared Thitarodes larvae, host of the Chinese medicinal fungus Ophiocordyceps sinensis on Tibetan plateau. Insects 2021, 12, 327. [Google Scholar] [CrossRef]
- Oliveira, J.M.M.; Henriques, I.; Read, D.S.; Gweon, H.S.; Morgado, R.G.; Peixoto, S.; Correia, A.; Soares, A.M.V.M.; Loureiro, S. Gut and faecal bacterial community of the terrestrial isopod Porcellionides pruinosus: Potential use for monitoring exposure scenarios. Ecotoxicology 2021, 30, 2096–2108. [Google Scholar] [CrossRef]
- Sela, R.; Laviad-Shitrit, S.; Halpern, M. Changes in microbiota composition along the metamorphosis developmental stages of Chironomus transvaalensis. Front. Microbiol. 2020, 11, 586678. [Google Scholar] [CrossRef]
- Xue, H.; Zhu, X.; Wang, L.; Zhang, K.; Li, D.; Ji, J.; Niu, L.; Wu, C.; Gao, X.; Luo, J.; et al. Gut bacterial diversity in different life cycle stages of Adelphocoris suturalis (Hemiptera: Miridae). Front. Microbiol. 2021, 12, 670383. [Google Scholar] [CrossRef] [PubMed]
- Franzini, P.Z.; Ramond, J.B.; Scholtz, C.H.; Sole, C.L.; Ronca, S.; Cowan, D.A. The gut microbiomes of two Pachysoma macLeay desert dung beetle species (Coleoptera: Scarabaeidae: Scarabaeinae) feeding on different diets. PLoS ONE 2016, 11, e0161118. [Google Scholar]
- Wang, Y.; Gilbreath, T.M., 3rd; Kukutla, P.; Yan, G.; Xu, J. Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya. PLoS ONE 2011, 6, e24767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colman, D.R.; Toolson, E.C.; Takacs-Vesbach, C.D. Do diet and taxonomy influence insect gut bacterial communities? Mol. Ecol. 2012, 21, 5124–5137. [Google Scholar] [CrossRef] [PubMed]
- Bozorov, T.A.; Rasulov, B.A.; Zhang, D. Characterization of the gut microbiota of invasive Agrilus mali Matsumara (Coleoptera: Buprestidae) using high-throughput sequencing: Uncovering plant cell-wall degrading bacteria. Sci. Rep. 2019, 9, 4923. [Google Scholar] [CrossRef] [Green Version]
- Idowua, A.B.; Edemaa, M.O.; Oyedepoa, M.T. Extracellular enzyme production by microflora from the gut region of the variegated grasshopper Zonocerus variegatus (Orthoptera: Pyrgomorphidae). Int. J. Trop. Insect Sci. 2009, 29, 229–235. [Google Scholar] [CrossRef]
- Lima, M.S.; Laport, M.S.; Lorosa, E.S.; Jurberg, J.; Dos Santos, K.R.N.; da Silva Neto, M.A.C.; Rachid, C.; Atella, G.C. Bacterial community composition in the salivary glands of triatomines (Hemiptera: Reduviidae). PLoS Negl. Trop. Dis. 2018, 12, e0006739. [Google Scholar] [CrossRef] [Green Version]
- Sabree, Z.L.; Huang, C.Y.; Arakawa, G.; Tokuda, G.; Lo, N.; Watanabe, H.; Moran, N.A. Genome shrinkage and loss of nutrient-providing potential in the obligate symbiont of the primitive termite Mastotermes darwiniensis. Appl. Environ. Microbiol. 2012, 78, 204–210. [Google Scholar] [CrossRef] [Green Version]
- Schauer, C.; Thompson, C.; Brune, A. Pyrotag sequencing of the gut microbiota of the cockroach Shelfordella lateralis reveals a highly dynamic core but only limited effects of diet on community structure. PLoS ONE 2014, 9, e85861. [Google Scholar] [CrossRef]
- Andongma, A.A.; Wan, L.; Dong, Y.C.; Wang, Y.L.; He, J.; Niu, C.Y. Assessment of the bacteria community structure across life stages of the Chinese citrus fly, Bactrocera minax (Diptera: Tephritidae). BMC Microbiol. 2019, 19, 285. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.K.; Ye, K.T.; Huang, W.F.; Ying, B.H.; Su, X.; Lin, L.H.; Li, J.H.; Chen, Y.P.; Li, J.L.; Bao, X.L.; et al. Influence of feeding type and Nosema ceranae infection on the gut microbiota of Apis cerana workers. mSystems 2018, 3, e00177-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, J.G.; Powell, S.; Kronauer, D.J.; Vasconcelos, H.L.; Frederickson, M.E.; Pierce, N.E. Stability and phylogenetic correlation in gut microbiota: Lessons from ants and apes. Mol. Ecol. 2014, 23, 1268–1283. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.E.; Russell, J.A.; Moreau, C.S.; Kautz, S.; Sullam, K.E.; Hu, Y.; Basinger, U.; Mott, B.M.; Buck, N.; Wheeler, D.E. Highly similar microbial communities are shared among related and trophically similar ant species. Mol. Ecol. 2012, 21, 2282–2296. [Google Scholar] [CrossRef]
- Curtis, T.P.; Sloan, W.T. Prokaryotic diversity and its limits: Microbial community structure in nature and implications for microbial ecology. Curr. Opin. Microbiol. 2004, 7, 221–226. [Google Scholar] [CrossRef]
- Sogin, M.L.; Morrison, H.G.; Huber, J.A.; Mark Welch, D.; Huse, S.M.; Neal, P.R.; Arrieta, J.M.; Herndl, G.J. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl. Acad. Sci. USA 2006, 103, 12115–12120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Serrano, F.; Perez-Cobas, A.E.; Rosas, T.; Baixeras, J.; Latorre, A.; Moya, A. The gut microbiota composition of the moth Brithys crini reflects insect metamorphosis. Microb. Ecol. 2019, 79, 960–970. [Google Scholar] [CrossRef]
- Zhukova, M.; Sapountzis, P.; Schiott, M.; Boomsma, J.J. Diversity and transmission of gut bacteria in atta and acromyrmex leaf-cutting ants during development. Front. Microbiol. 2017, 8, 1942. [Google Scholar] [CrossRef]
- Zurek, K.; Nayduch, D. Bacterial associations across house fly life history: Evidence for transstadial carriage from managed manure. J. Insect Sci. 2016, 16, 2. [Google Scholar] [CrossRef]
- Mohlmann, T.W.R.; Ter Braak, C.J.F.; Te Beest, D.E.; Hendriks, M.; Nijhuis, E.H.; Warris, S.; Drolet, B.S.; van Overbeek, L.; Koenraadt, C.J.M. Species identity, life history, and geographic distance influence gut bacterial communities in lab-reared and European field-collected Culicoides biting midges. Microb. Ecol. 2021. Epub ahead of print. [Google Scholar] [CrossRef]
- Hadapad, A.B.; Shettigar, S.K.G.; Hire, R.S. Bacterial communities in the gut of wild and mass-reared Zeugodacus cucurbitae and Bactrocera dorsalis revealed by metagenomic sequencing. BMC Microbiol. 2019, 19, 282. [Google Scholar] [CrossRef]
- Wang, A.; Yao, Z.; Zheng, W.; Zhang, H. Bacterial communities in the gut and reproductive organs of Bactrocera minax (Diptera: Tephritidae) based on 454 pyrosequencing. PLoS ONE 2014, 9, e106988. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.Y.; Cui, Y.H.; Chu, X.; Li, G.Q.; Yang, M.J.; Wang, R.; Liang, G.H.; Wu, S.Q.; Tigabu, M.; Zhang, F.P.; et al. Gut bacterial communities of Lymantria xylina and their associations with host development and diet. Microorganisms 2021, 9, 1860. [Google Scholar] [CrossRef] [PubMed]
- Lauzon, C.R.; McCombs, S.D.; Potter, S.E.; Peabody, N.C. Establishment and vertical passage of Enterobacter (Pantoea) Agglomerans and Klebsiella pneumoniae through all life stages of the Mediterranean fruit fly (Diptera: Tephritidae). Ann. Entomol. Soc. Am. 2009, 102, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Behar, A.; Jurkevitch, E.; Yuval, B. Bringing back the fruit into fruit fly-bacteria interactions. Mol. Ecol. 2008, 17, 1375–1386. [Google Scholar] [CrossRef] [PubMed]
- Rosas, T.; Garcia-Ferris, C.; Dominguez-Santos, R.; Llop, P.; Latorre, A.; Moya, A. Rifampicin treatment of Blattella germanica evidences a fecal transmission route of their gut microbiota. FEMS Microbiol. Ecol. 2018, 94, fiy002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salem, H.; Florez, L.; Gerardo, N.; Kaltenpoth, M. An out-of-body experience: The extracellular dimension for the transmission of mutualistic bacteria in insects. Proc. Biol. Sci. 2015, 282, 20142957. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Fernandez, S.; Mazzoni, V.; Pedrazzoli, F.; Pertot, I.; Campisano, A. A phloem-feeding insect transfers bacterial endophytic communities between grapevine plants. Front. Microbiol. 2017, 8, 834. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Sex | Developmental Stage | Time Points |
|---|---|---|---|
| MN12 | Male | Early nymphs | 1st instar male nymphs, MN1 |
| 2nd instar male nymphs, MN2 | |||
| MN35 | Male | Middle nymphs | 3rd instar male nymphs, MN3 |
| 4th instar male nymphs, MN4 | |||
| 5th instar male nymphs, MN5 | |||
| MN67 | Male | Late nymphs | 6th instar male nymphs, MN6 |
| 7th instar male nymphs, MN7 | |||
| MA | Male | Adults | 0 day male adult, MA1 |
| 7 day male adult, MA2 | |||
| 14 day male adult, MA3 | |||
| 21 day male adult, MA4 | |||
| FN12 | Female | Early nymphs | 1st instar female nymphs, FN1 |
| 2nd instar female nymphs, FN2 | |||
| FN35 | Female | Middle nymphs | 3rd instar female nymphs, FN3 |
| 4th instar female nymphs, FN4 | |||
| 5th instar female nymphs, FN5 | |||
| FN67 | Female | Late nymphs | 6th instar female nymphs, FN6 |
| 7th instar female nymphs, FN7 | |||
| FA | Female | Adults | 0 day female adult, FA1 |
| 7 day female adult, FA2 | |||
| 14 day female adult, FA3 | |||
| 21 day female adult, FA4 |
| Taxonomic Levels | No. OTUs (%) | % Read Counts |
|---|---|---|
| Proteobacteria | 2208 (74.04%) | 57.22% |
| Gammaproteobacteria | 2083 (69.85%) | 57.05% |
| Enterobacteriales | 2000 (67.07%) | 56.39% |
| Enterobacteriaceae | 2000 (67.07%) | 56.39% |
| Kluyvera | 387 (12.98%) | 14.62% |
| Obesumbacterium | 339 (11.37%) | 0.26% |
| Buttiauxella | 296 (9.93%) | 1.01% |
| Hafnia | 152 (5.10%) | 33.17% |
| Serratia | 75 (2.52%) | 1.98% |
| Raoultella | 44 (1.48%) | 0.16% |
| Alphaproteobacteria | 57 (1.91%) | 0.08% |
| Betaproteobacteria | 35 (1.17%) | 0.08% |
| Firmicutes | 511 (17.14%) | 42.59% |
| Bacilli | 418 (14.02%) | 42.52% |
| Lactobacillales | 367 (12.31%) | 41.83% |
| Lactobacillaceae | 300 (10.06%) | 41.34% |
| Lactobacillus | 286 (9.59%) | 40.71% |
| Clostridia | 81 (2.72%) | 0.06% |
| Bacteroidetes | 58 (1.95%) | 0.04% |
| Bacteroidia | 41 (1.37%) | 0.03% |
| Acidobacteria | 55 (1.84%) | 0.01% |
| OTU ID | % Read Counts | Phylum | Class | Order | Family | Genus |
|---|---|---|---|---|---|---|
| Otu128 | 0.14% | Firmicutes | Bacilli | Lactobacillales | Enterococcaceae | Enterococcus |
| Otu2 | 38.95% | Firmicutes | Bacilli | Lactobacillales | Lactobacillaceae | Lactobacillus |
| Otu408509 | 0.04% | Firmicutes | Bacilli | Lactobacillales | Lactobacillaceae | Lactobacillus |
| Otu95456 | 0.02% | Firmicutes | Bacilli | Lactobacillales | Lactobacillaceae | Lactobacillus |
| Otu211 | 0.09% | Firmicutes | Bacilli | Lactobacillales | Leuconostocaceae | Weissella |
| Otu125 | 0.18% | Firmicutes | Bacilli | Lactobacillales | Streptococcaceae | Lactococcus |
| Otu107 | 0.11% | Firmicutes | Bacilli | Lactobacillales | Streptococcaceae | Lactococcus |
| Otu92 | 0.04% | Firmicutes | Bacilli | Lactobacillales | Streptococcaceae | Lactococcus |
| Otu212 | 0.06% | Proteobacteria | Betaproteobacteria | Burkholderiales | Burkholderiaceae | Burkholderia |
| Otu14025 | 0.21% | Proteobacteria | Gammaproteobacteria | Enterobacteriales | Enterobacteriaceae | Buttiauxella |
| Otu66 | 1.48% | Proteobacteria | Gammaproteobacteria | Enterobacteriales | Enterobacteriaceae | Citrobacter |
| Otu6 | 0.82% | Proteobacteria | Gammaproteobacteria | Enterobacteriales | Enterobacteriaceae | Escherichia/Shigella |
| Otu1 | 13.90% | Proteobacteria | Gammaproteobacteria | Enterobacteriales | Enterobacteriaceae | Hafnia |
| Otu8 | 10.12% | Proteobacteria | Gammaproteobacteria | Enterobacteriales | Enterobacteriaceae | Hafnia |
| Otu10 | 7.70% | Proteobacteria | Gammaproteobacteria | Enterobacteriales | Enterobacteriaceae | Hafnia |
| Otu3 | 15.26% | Proteobacteria | Gammaproteobacteria | Enterobacteriales | Enterobacteriaceae | Kluyvera |
| Otu22 | 0.65% | Proteobacteria | Gammaproteobacteria | Enterobacteriales | Enterobacteriaceae | Serratia |
| Otu27 | 0.87% | Proteobacteria | Gammaproteobacteria | Enterobacteriales | Enterobacteriaceae | Unclassified |
| Otu210 | 0.41% | Proteobacteria | Gammaproteobacteria | Enterobacteriales | Enterobacteriaceae | Unclassified |
| Otu123 | 0.07% | Proteobacteria | Gammaproteobacteria | Pseudomonadales | Pseudomonadaceae | Pseudomonas |
| Otu95457 | 0.02% | Thermotogae | Thermotogae | Petrotogales | Petrotogaceae | Defluviitoga |
| Group | Sample Size | Number of OTUs | Chao1 | ACE | Shannon | Simpson |
|---|---|---|---|---|---|---|
| MN12 | 6 | 475.667 ± 109.432 | 710.841 ± 159.337 | 722.539 ± 174.095 | 1.568 ± 0.383 | 0.667 ± 0.103 |
| MN35 | 9 | 459.333 ± 40.765 | 682.463 ± 61.026 | 705.487 ± 78.775 | 1.602 ± 0.262 | 0.659 ± 0.078 |
| MN67 | 6 | 396.667 ± 78.194 | 659.140 ± 78.934 | 664.879 ± 83.689 | 1.567 ± 0.403 | 0.644 ± 0.168 |
| MA | 12 | 355.500 ± 153.390 | 565.359 ± 193.420 | 582.558 ± 183.128 | 1.620 ± 0.318 | 0.683 ± 0.077 |
| FN12 | 6 | 512.000 ± 83.816 | 720.064 ± 161.132 | 736.850 ± 163.78 | 1.927 ± 0.275 | 0.760 ± 0.084 |
| FN35 | 9 | 419.444 ± 61.561 | 615.366 ± 110.787 | 647.467 ± 119.32 | 1.577 ± 0.418 | 0.632 ± 0.185 |
| FN67 | 6 | 592.000 ± 179.861 | 803.358 ± 194.458 | 821.251 ± 202.958 | 1.707 ± 0.430 | 0.663 ± 0.163 |
| FA | 12 | 446.833 ± 157.623 | 663.298 ± 163.211 | 667.523 ± 173.058 | 1.812 ± 0.408 | 0.714 ± 0.134 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Z.; Huang, H.; Che, X. Bacterial Communities in the Feces of Laboratory Reared Gampsocleis gratiosa (Orthoptera: Tettigoniidae) across Different Developmental Stages and Sexes. Insects 2022, 13, 361. https://doi.org/10.3390/insects13040361
Zhou Z, Huang H, Che X. Bacterial Communities in the Feces of Laboratory Reared Gampsocleis gratiosa (Orthoptera: Tettigoniidae) across Different Developmental Stages and Sexes. Insects. 2022; 13(4):361. https://doi.org/10.3390/insects13040361
Chicago/Turabian StyleZhou, Zhijun, Huimin Huang, and Xuting Che. 2022. "Bacterial Communities in the Feces of Laboratory Reared Gampsocleis gratiosa (Orthoptera: Tettigoniidae) across Different Developmental Stages and Sexes" Insects 13, no. 4: 361. https://doi.org/10.3390/insects13040361