Transcriptome Sequencing Highlights the Regulatory Role of DNA Methylation in Immune-Related Genes’ Expression of Chinese Oak Silkworm, Antheraea pernyi


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Insects
2.2. 5-Azacytidine Administration, Bacterial Challenge, and Tissue Collection
2.3. RNA Preparation, Library Construction, and Sequencing
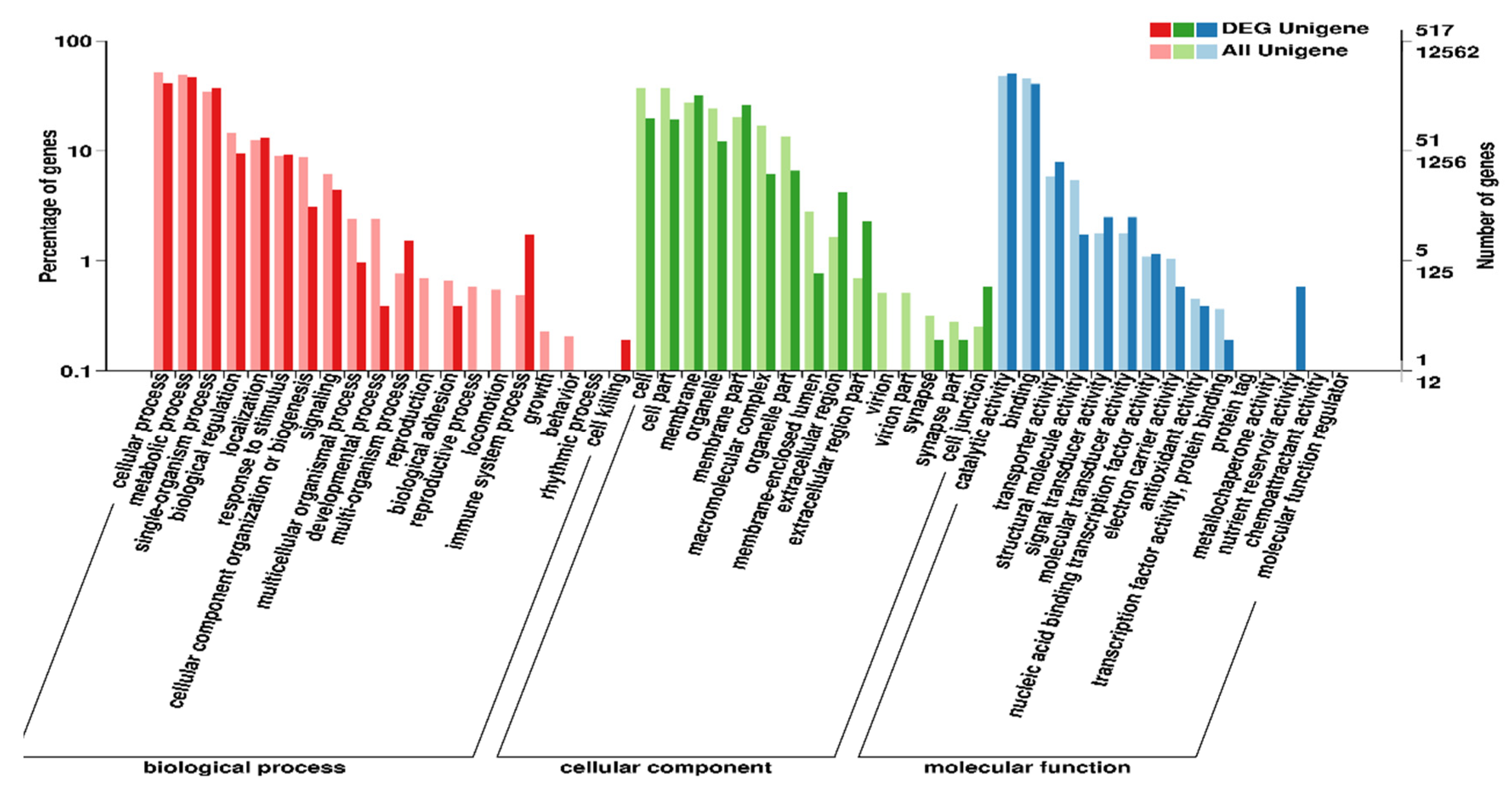
2.4. Transcriptome Assembly, Annotation, and Function Enrichment
2.5. Unigene Annotation and Functional Classification
2.6. Quantitative RT-PCR Analysis
2.7. Statistical Analysis
3. Results
3.1. Summary of Transcriptome Sequence and Assembly
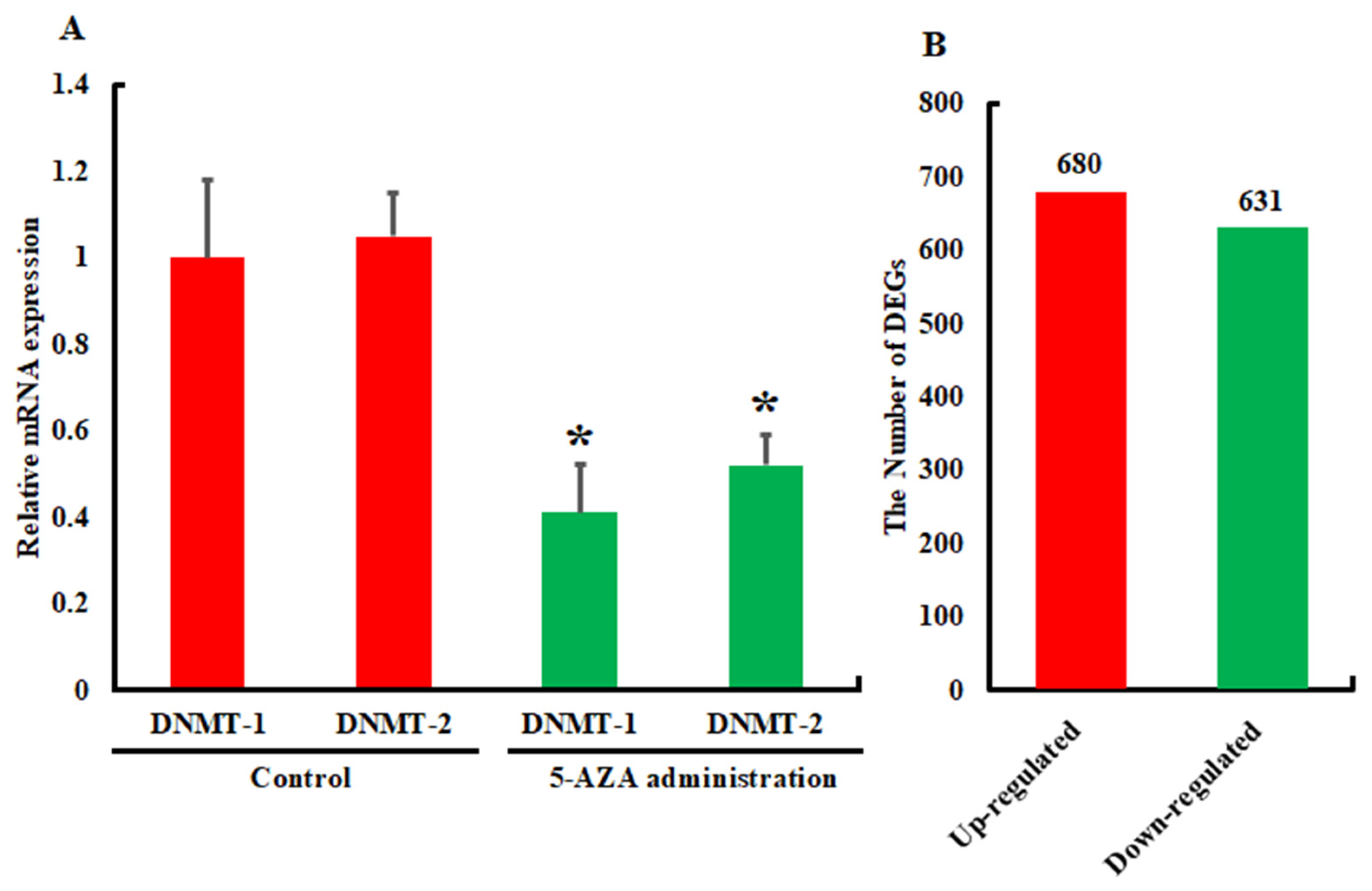
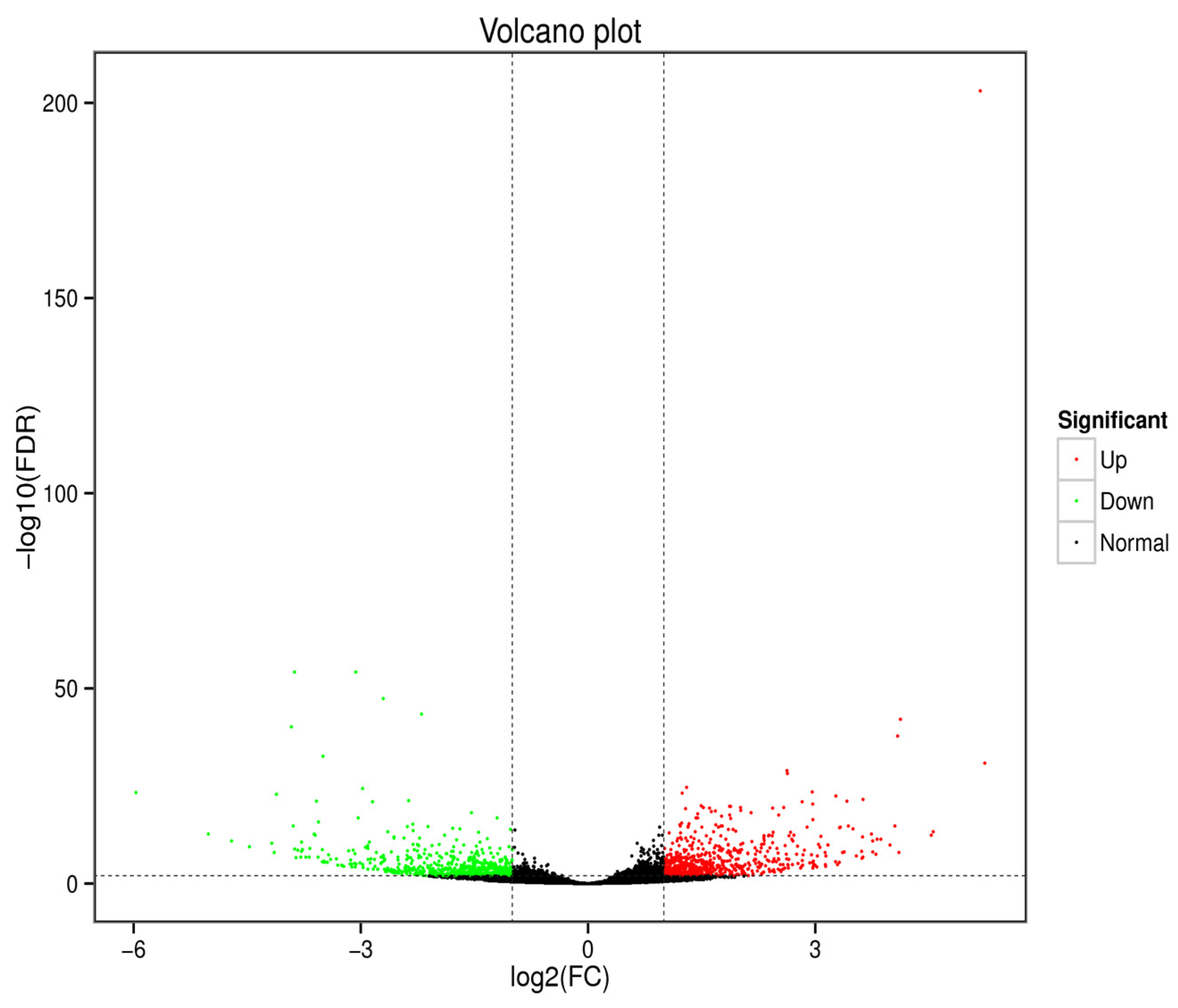
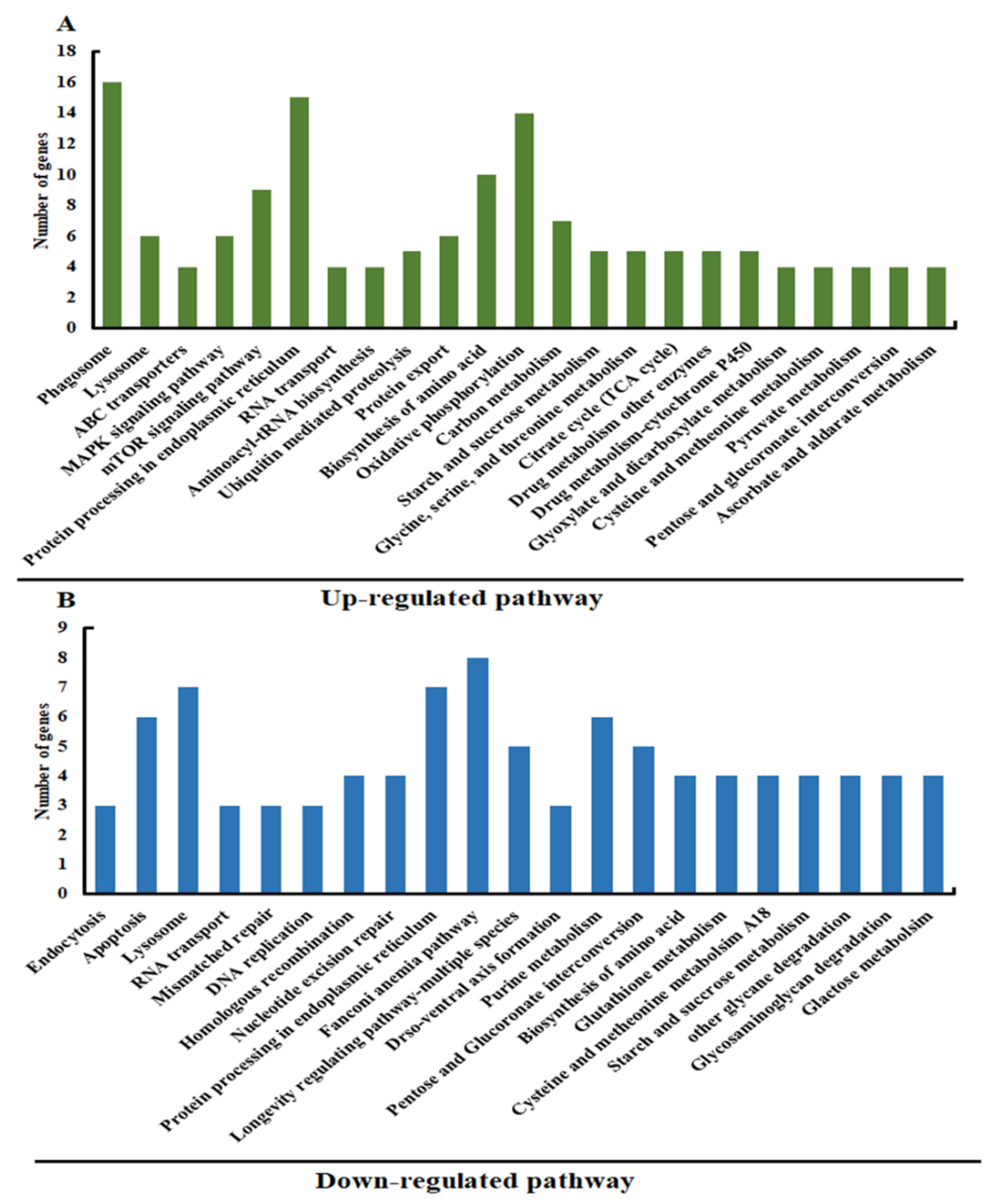
3.2. Analysis of Differentially Expressed Genes
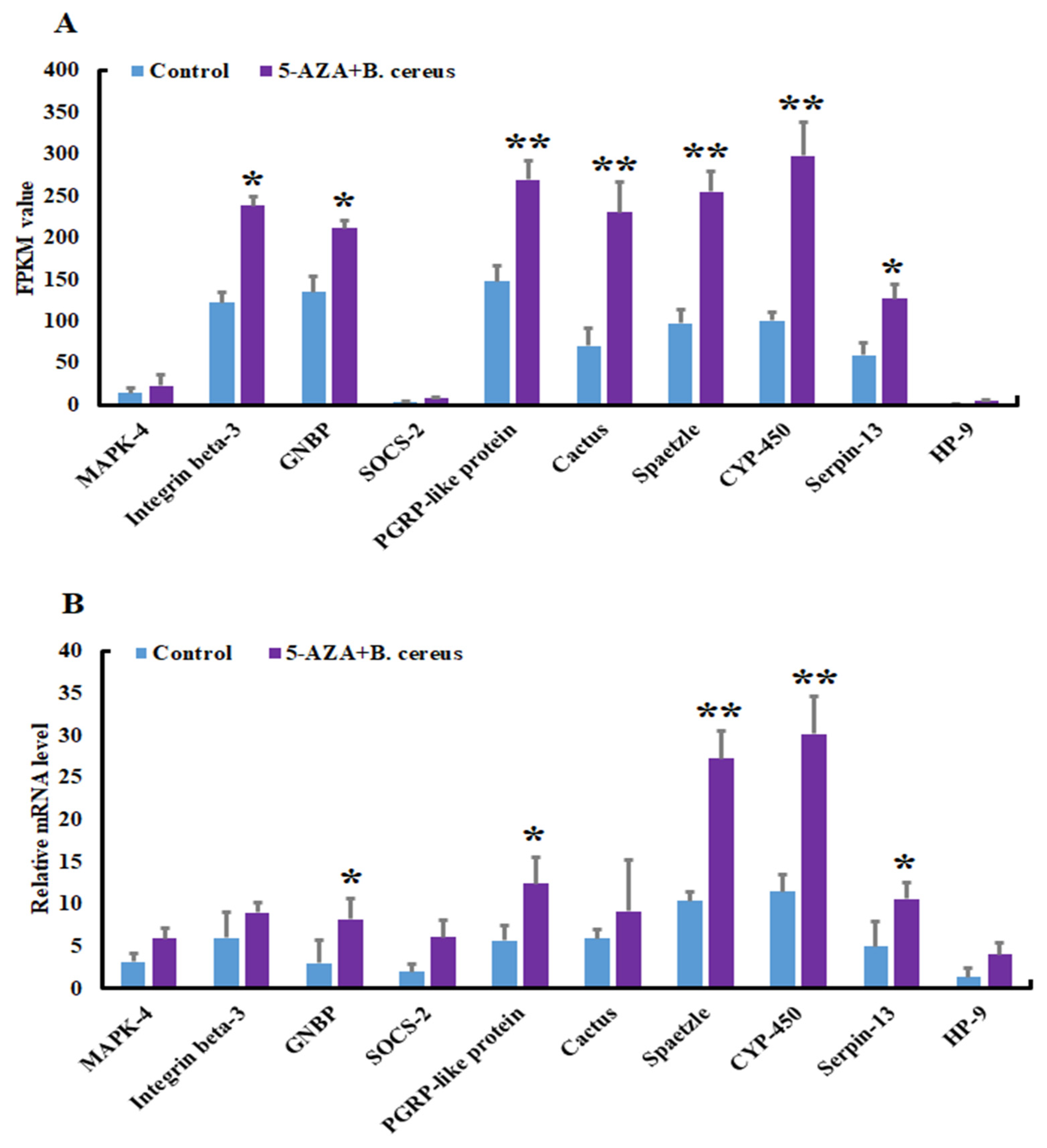
3.3. Validation and Reliability of the Transcriptome Data by qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Park, J.; Peng, Z.; Zeng, J.; Elango, N.; Park, T.; Wheeler, D.; Werren, J.H.; Yi, S.V. Comparative analyses of DNA methylation and sequence evolution using Nasonia genomes. Mol. Biol. Evol. 2011, 28, 3345–3354. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome-biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Riccio, A.; Alvania, R.S.; Lonze, B.E.; Ramanan, N.; Kim, T.; Huang, Y.; Dawson, T.M.; Snyder, S.H.; Ginty, D.D. A nitric oxide signaling pathway controls CREB-mediated gene expression in neurons. Mol. Cell 2009, 21, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Cicchini, L.; Blumhagen, R.Z.; Westrich, J.A.; Myers, M.E.; Warren, C.J.; Siska, C.; Raben, D.; Kechris, K.J.; Pyeon, D. High-risk human papillomavirus E7 alters host DNA Methylome and represses HLA-E expression in human keratinocytes. Sci. Rep. 2017, 7, 3633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dongt, S.M.; Lee, H.G.; Cho, S.G.; Kwon, S.H.; Yoon, H.; Kwon, H.J.; Lee, J.H.; Kim, H.; Park, P.G.; Kim, H.; et al. Hypermethylation of the interferon regulatory factor 5 promoter in Epstein-Barr virus-associated gastric carcinoma. J. Microbiol. 2015, 53, 70–76. [Google Scholar] [CrossRef]
- Di Bartolo, D.L.; Cannon, M.; Liu, Y.F.; Renne, R.; Chadburn, A.; Boshoff, C.; Cesarman, E. KSHV LANA inhibits TGF-beta signaling through epigenetic silencing of the TGF-beta type II receptor. Blood 2008, 111, 4731–4740. [Google Scholar] [CrossRef]
- Kang, M.S.; Kieff, E. Epstein-Barr virus latent genes. Exp. Mol. Med. 2015, 47, e131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Xu, Y.; Mo, D.; Huang, P.; Sun, R.; Huang, L.; Pan, S.; Xu, J. Kaposi’s sarcoma-associated herpesvirus (KSHV) vIL-6 promotes cell proliferation and migration by upregulating DNMT1 via STAT3 activation. PLoS ONE 2014, 9, e93478. [Google Scholar] [CrossRef]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating agents target colorectal Cancer cells by inducing viral mimicry by endogenous transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA methylation causes an interferon response in Cancer via dsRNA including endogenous retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [Green Version]
- Capuano, F.; Mulleder, M.; Kok, R.; Blom, H.J.; Ralser, M. Cytosine DNA methylation is found in Drosophila melanogaster but absent in Saccharomyces cerevisiae, Schizosaccharomyces pombe, and other yeast species. Anal. Chem. 2014, 86, 3697–3702. [Google Scholar] [CrossRef]
- Xiang, H.; Zhu, J.; Chen, Q.; Dai, F.; Li, X.; Li, M.; Zhang, H.; Zhang, G.; Li, D.; Dong, Y.; et al. Single base-resolution methylome of the silkworm reveals a sparse epigenomic map. Nat. Biotechnol. 2010, 28, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Feliciello, I.; Parazajder, J.; Akrap, I.; Ugarkovic, D. First evidence of DNA methylation in insect Tribolium castaneum: Environmental regulation of DNA methylation within heterochromatin. Epigenetics 2013, 8, 534–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beeler, S.M.; Wong, G.T.; Zheng, J.M.; Bush, E.C.; Remnant, E.J.; Oldroyd, B.P.; Drewell, R.A. Whole-genome DNA methylation profile of the jewel wasp (Nasonia vitripennis). G3 (Bethesda) 2014, 4, 383–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glastad, K.M.; Hunt, B.G.; Yi, S.V.; Goodisman, M.A. DNA methylation in insects: On the brink of the epigenomic era. Insect Mol. Biol. 2011, 20, 553–565. [Google Scholar] [CrossRef]
- Standage, D.S.; Berens, A.J.; Glastad, K.M.; Severin, A.J.; Brendel, V.P.; Toth, A.L. Genome, transcriptome and methylome sequencing of a primitively eusocial wasp reveal a greatly reduced DNA methylation system in a social insect. Mol. Ecol. 2016, 25, 1769–1784. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Jie, W.; Shang, Q.; Annan, E.; Jiang, X.; Hou, C.; Chen, T.; Guo, X. DNA methylation in silkworm genome may provide insights into epigenetic regulation of response to Bombyx mori cypovirus infection. Sci. Rep. 2017, 7, 16013. [Google Scholar] [CrossRef] [Green Version]
- Bonasio, R.; Li, Q.; Lian, J.; Mutti, N.S.; Jin, L.; Zhao, H.; Zhang, P.; Wen, P.; Xiang, H.; Ding, Y.; et al. Genome-wide and caste-specific DNA methylomes of the ants Camponotus floridanus and Harpegnathos saltator. Curr. Biol. 2012, 22, 1755–1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Bonasio, R.; Simola, D.F.; Liebig, J.; Berger, S.L.; Reinberg, D. DNA methylation in social insects: How epigenetics can control behavior and longevity. Annu. Rev. Entomol. 2015, 60, 435–452. [Google Scholar] [CrossRef] [PubMed]
- Biergans, S.D.; Giovanni, G.C.; Judith, R.; Charles, C. Dnmts and Tettarget memory-associated genes after appetitive olfactory training in honey bees. Sci. Rep. 2015, 5, 21656. [Google Scholar] [CrossRef] [Green Version]
- Herb, B.R.; Wolschin, F.; Hansen, K.D.; Aryee, M.J.; Langmead, B.; Irizarry, R.; Amdam, G.V.; Feinberg, A.P. Reversible switching between epigenetic states in honeybee behavioral subcastes. Nat. Neurosci. 2012, 15, 1371–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kausar, S.; Abbas, M.N.; Cui, H. A review on the DNA methyltransferase family of insects: Aspect and prospects. Int. J. Biol. Macromol. 2021, 186, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Baradaran, E.; Moharramipour, S.; Asgari, S.; Mehrabadi, M. Induction of DNA methyltransferase genes in Helicoverpa armigera following injection of pathogenic bacteria modulates expression of antimicrobial peptides and affects bacterial proliferation. J. Insect Physiol. 2019, 118, 103939. [Google Scholar] [CrossRef]
- Heitmueller, M.; Billion, A.; Dobrindt, U.; Vilcinskas, A.; Mukherjee, K. Epigenetic mechanisms regulate innate immunity against uropathogenic and commensal-like Escherichia coli in the surrogate insect model Galleria mellonella. Infect. Immun. 2017, 85, e00336-17. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.H.; Woolfit, M.; Huttley, G.A.; Rances, E.; Caragata, E.P.; Popovici, J.; O’Neill, S.L.; McGraw, E.A. Infection with a virulent strain of Wolbachia disrupts genome wide-patterns of cytosine methylation in the mosquito Aedes aegypti. PLoS ONE 2013, 8, e66482. [Google Scholar] [CrossRef] [PubMed]
- Vilcinkas, A. The role of epigenetics in host–parasite coevolution: Lessons from the model host insects galleria mellonella and Tribolium castaneum. Zoology 2016, 119, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Xin, Z.Z.; Liu, Q.N.; Liu, Y.; Zhang, D.Z.; Wang, Z.F.; Zhang, H.B.; Ge, B.M.; Zhou, C.L.; Chai, X.Y.; Tang, B.P. Transcriptome-Wide Identification of Differentially Expressed Genes in Chinese Oak Silkworm Antheraea pernyi in Response to Lead Challenge. J. Agric. Food Chem. 2017, 65, 9305–9314. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Kunert, N.; Marhold, J.; Stanke, J.; Stach, D.; Lyko, F. A Dnmt2-like protein mediates DNA methylation in Drosophila. Development 2003, 130, 5083–5090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, O.; Siman-Tov, R.; Ankri, S. Characterization of cytosine methylated regions and 5-cytosine DNA methyltransferase (Ehmeth) in the protozoan parasite Entamoeba histolytica. Nucleic Acids Res. 2004, 32, 287–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, M.; Curk, T.; Xu, Q.; Zupan, B.; Kuspa, A.; Shaulsky, G. Developmentally regulated DNA methylation in Dictyostelium discoideum. Eukaryot. Cell 2006, 5, 18–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Wang, Y.F.; Cantemir, C.; Muller, M.T. Endogenous assays of DNA methyltransferases: Evidence for differential activities of DNMT1, DNMT2, and DNMT3 in mammalian cells In vivo. Mol. Cell. Biol. 2003, 23, 2709–2719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, M.; Hagemann, S.; Hanna, K.; Lyko, F. Azacytidine inhibits RNA methylation at DNMT2 target sites in human cancer cell lines. Cancer Res. 2009, 69, 8127–8132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geyer, K.K.; Rodríguez López, C.M.; Chalmers, I.W.; Munshi, S.E.; Truscott, M.; Heald, J.; Wilkinson, M.J.; Hoffmann, K.F. Cytosine methylation regulates oviposition in the pathogenic blood fluke Schistosoma mansoni Kathrin. Nat. Commun. 2011, 2, 424. [Google Scholar] [CrossRef] [Green Version]
- Walderdorff, L.; Laval-Gilly, P.; Bonnefoy, A.; Falla-Angel, J. Imidacloprid intensifies its impact on honeybee and bumblebee cellular immune response when challenged with LPS (lippopolysacharide) of Escherichia coli. J. Insect Physiol. 2018, 108, 17–24. [Google Scholar] [CrossRef]
- Kausar, S.; Abbas, M.N.; Zhao, Y.; Cui, H. Immune strategies of silkworm, Bombyx mori against microbial infections. Invertebr. Surviv. J. 2019, 16, 130–140. [Google Scholar]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- De Monerri, N.C.S.; Kim, K. Pathogens hijack the epigenome: A new twist on host-pathogen interactions. Am. J. Pathol. 2014, 184, 897–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGrath-Morrow, S.A.; Ndeh, R.; Helmin, K.A.; Chen, S.Y.; Anekalla, K.R.; Abdala-Valencia, H.; D’Alessio, F.R.; Collaco, J.M.; Singer, B.D. DNA methylation regulates the neonatal CD4+ T-cell response to pneumonia in mice. J. Biol. Chem. 2018, 293, 11772–11783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurson, J.; Khan, S.; Chung, R.; Cross, K.; Raj, K. Epigenetic repression of E-cadherin by human papillomavirus 16 E7 protein. Carcinogenesis 2010, 31, 918–926. [Google Scholar] [CrossRef] [Green Version]
- Tolg, C.; Sabha, N.; Cortese, R.; Panchal, T.; Ahsan, A.; Soliman, A.; Aitken, K.J.; Petronis, A.; Bägli, D.J. Uropathogenic, E. coli infection provokes epigenetic downregulation of CDKN2A (p16INK4A) in uroepithelial cells. Lab Investig. 2011, 91, 825–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacis, A.; Tailleux, L.; Morin, A.M.; Lambourne, J.; MacIsaac, J.L.; Yotova, V.; Dumaine, A.; Danckaert, A.; Luca, F.; Grenier, J.C.; et al. Bacterial infection remodels the DNA methylation landscape of human dendritic cells. Genome Res. 2015, 25, 1801–1811. [Google Scholar] [CrossRef] [Green Version]
- Lemaitre, B.; Nicolas, E.; Michaut, L.; Reichhart, J.M.; Hoffmann, J.A. The dorsoventral regulatory gene cassette spätzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 1996, 86, 973–983. [Google Scholar] [CrossRef] [Green Version]
- Kajla, M.; Choudhury, T.P.; Kakani, P.; Gupta, K.; Dhawan, R.; Gupta, L.; Kumar, S. Silencing of Anopheles stephensi heme peroxidase HPX15 activates diverse immune pathways to regulate the growth of midgut bacteria. Front. Microbiol. 2016, 7, 1351. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Dhawan, R.; Kajla, M.; Misra, T.; Kumar, S.; Gupta, L. The evolutionary divergence of STAT transcription factor in different Anopheles species. Gene 2017, 596, 89–97. [Google Scholar] [CrossRef]
- Yu, B.; Sang, Q.; Pan, G.; Li, C.; Zhou, Z. A Toll-Spätzle Pathway in the immune response of Bombyx mori. Insects 2020, 11, 586. [Google Scholar] [CrossRef]
- Kingsolver, M.B.; Huang, Z.; Hardy, R.W. Insect antiviral innate immunity: Pathways, effectors, and connections. J. Mol. Biol. 2013, 425, 4921–4936. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, J.A. The immune response of Drosophila. Nature 2003, 426, 33–38. [Google Scholar] [CrossRef]
- Abbas, M.N.; Kausar, S.; Zhao, E.; Cui, H. Suppressors of cytokine signaling proteins as modulators of development and innate immunity of insects. Dev. Comp. Immunol. 2020, 104, 103561. [Google Scholar] [CrossRef]
- Abbas, M.N.; Kausar, S.; Gul, I.; Ke, X.X.; Dong, Z.; Lu, X.; Cui, H. Suppressor of cytokine signalling 6 is a potential regulator of antimicrobial peptides in the Chinese oak silkworm, Antheraea pernyi. Mol. Immunol. 2021, 140, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Xiang, J. Signaling pathways regulating innate immune responses in shrimp. Fish Shellfish. Immunol. 2013, 34, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Hedengren-Olcott, M.; Olcott, M.C.; Mooney, D.T.; Ekengren, S.; Geller, B.L.; Taylor, B.J. Differential activation of the NF-kappaB-like factors Relish and Dif in Drosophila melanogaster by fungi and Gram-positive bacteria. J. Biol. Chem. 2004, 279, 21121–21127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavine, M.D.; Strand, M.R. Haemocytes from Pseudoplusia includes express multiple alpha and beta integrin subunits Insect. Mol. Biol. 2003, 12, 441–452. [Google Scholar]
- Li, C.; Zhang, K.; Pan, G.; Zhang, L.; Hu, X.; Zhao, G.; Deng, C.; Tan, M.; Li, C.; Xu, M.; et al. Bmintegrin β1: A broadly expressed molecule modulates the innate immune response of Bombyx mori. Dev. Comp. Immunol. 2021, 114, 103869. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clean Bases | Clean Reads | Mapped Reads | Mapped Ratio | Q20% | Q30% | GC (%) | |
|---|---|---|---|---|---|---|---|
| 5-AZA and bacteria injected | 8,970,065,914 | 30,212,355 | 23,677,535 | 78.37 | 97.80 | 93.81 | 43.95 |
| 5-AZA and bacteria injected | 7,182,585,608 | 24,056,953 | 19,384,242 | 80.58 | 97.53 | 93.41 | 43.00 |
| 5-AZA and bacteria injected | 7,613,303,876 | 25,478,892 | 20,665,639 | 81.11 | 97.50 | 93.34 | 42.21 |
| Only bacteria injected | 7,333,560,222 | 24,534,056 | 19,994,004 | 81.49 | 97.48 | 93.20 | 40.95 |
| Only bacteria injected | 7,653,093,914 | 26,678,456 | 20,316,290 | 79.12 | 97.98 | 94.21 | 42.52 |
| Only bacteria injected | 7,018,186,154 | 23,536,596 | 18,881,662 | 80.22 | 97.73 | 93.69 | 42.58 |
| Length Range | Transcript | Unigene |
|---|---|---|
| 300–500 | 35,064 (29.90%) | 26,435 (47.95%) |
| 500–1000 | 28,596 (24.38%) | 14,437 (26.19%) |
| 1000–2000 | 25,629 (21.85%) | 7620 (13.82%) |
| 2000+ | 27,981 (23.86%) | 6638 (12.04%) |
| Total Number | 117,272 | 55,131 |
| Total Length | 167,150,431 | 53,875,332 |
| N50 Length | 2357 | 1621 |
| Mean Length | 1425.323 | 977.2239 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kausar, S.; Liu, R.; Gul, I.; Abbas, M.N.; Cui, H. Transcriptome Sequencing Highlights the Regulatory Role of DNA Methylation in Immune-Related Genes’ Expression of Chinese Oak Silkworm, Antheraea pernyi. Insects 2022, 13, 296. https://doi.org/10.3390/insects13030296
Kausar S, Liu R, Gul I, Abbas MN, Cui H. Transcriptome Sequencing Highlights the Regulatory Role of DNA Methylation in Immune-Related Genes’ Expression of Chinese Oak Silkworm, Antheraea pernyi. Insects. 2022; 13(3):296. https://doi.org/10.3390/insects13030296
Chicago/Turabian StyleKausar, Saima, Ruochen Liu, Isma Gul, Muhammad Nadeem Abbas, and Hongjuan Cui. 2022. "Transcriptome Sequencing Highlights the Regulatory Role of DNA Methylation in Immune-Related Genes’ Expression of Chinese Oak Silkworm, Antheraea pernyi" Insects 13, no. 3: 296. https://doi.org/10.3390/insects13030296