Integration of Transcriptomic and Proteomic Analyses Reveals New Insights into the Regulation of Immune Pathways in Midgut of Samia ricini upon SariNPV Infection


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Transcripome Analysis
2.2.1. RNA Extraction, cDNA Library Construction and Sequencing
2.2.2. Data Analysis and Quality Control
2.2.3. Expression Analysis of Differential Expressed Genes
2.3. Proteomics Methods
2.3.1. Total Protein Extraction
2.3.2. Protein Testing
2.3.3. Identification and Quantification of Proteins
2.3.4. Expression Analysis of Differential Proteins
2.4. qRT-PCR Verification
3. Results
3.1. Transcriptome Analysis of Midgut Samples
3.1.1. Statistics and Identification of Differentially Expressed Genes
3.1.2. Enrichment Analysis of DEGs
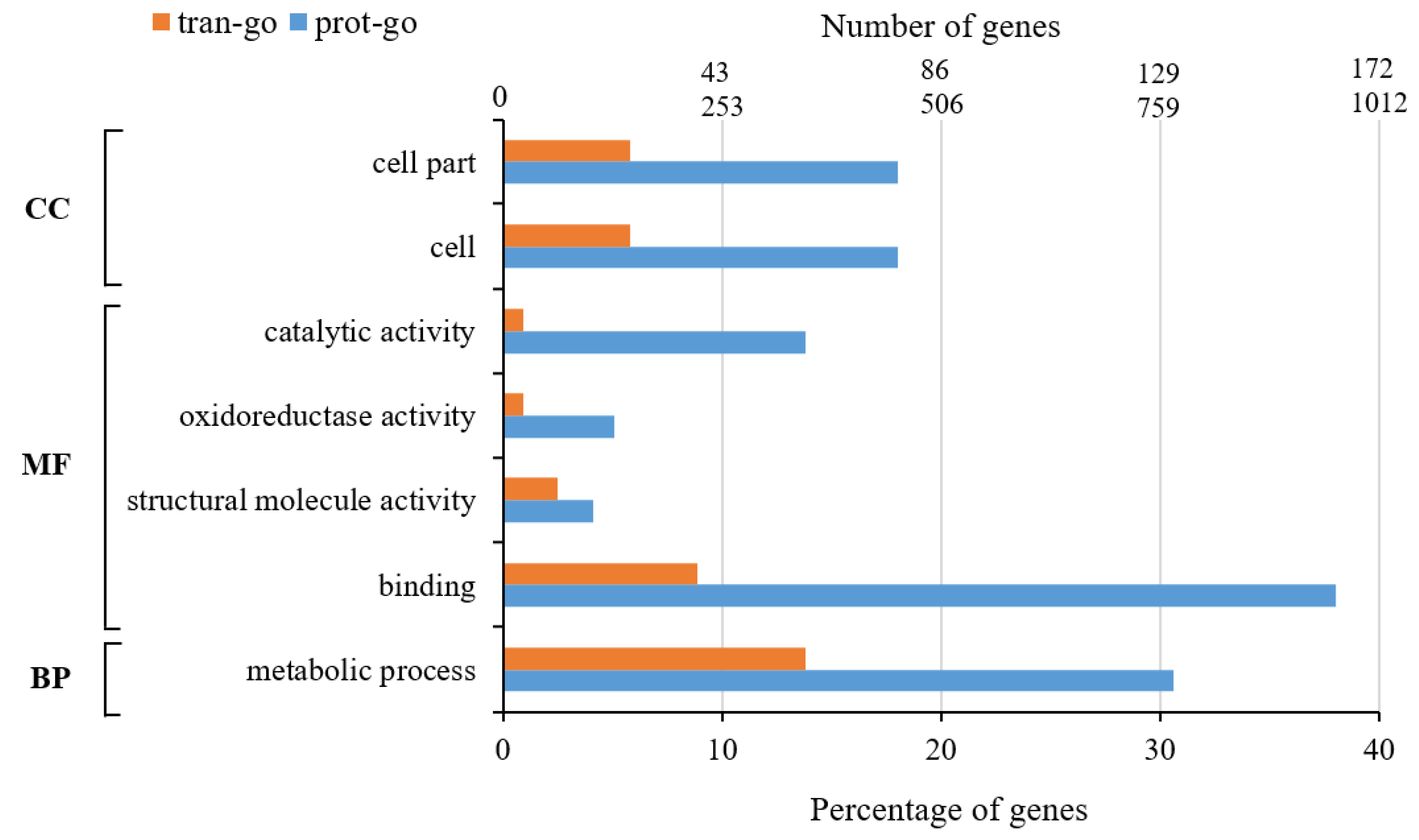
- Gene Ontology (GO) Function Annotation Analysis
- 2.
- KEGG function annotation analysis
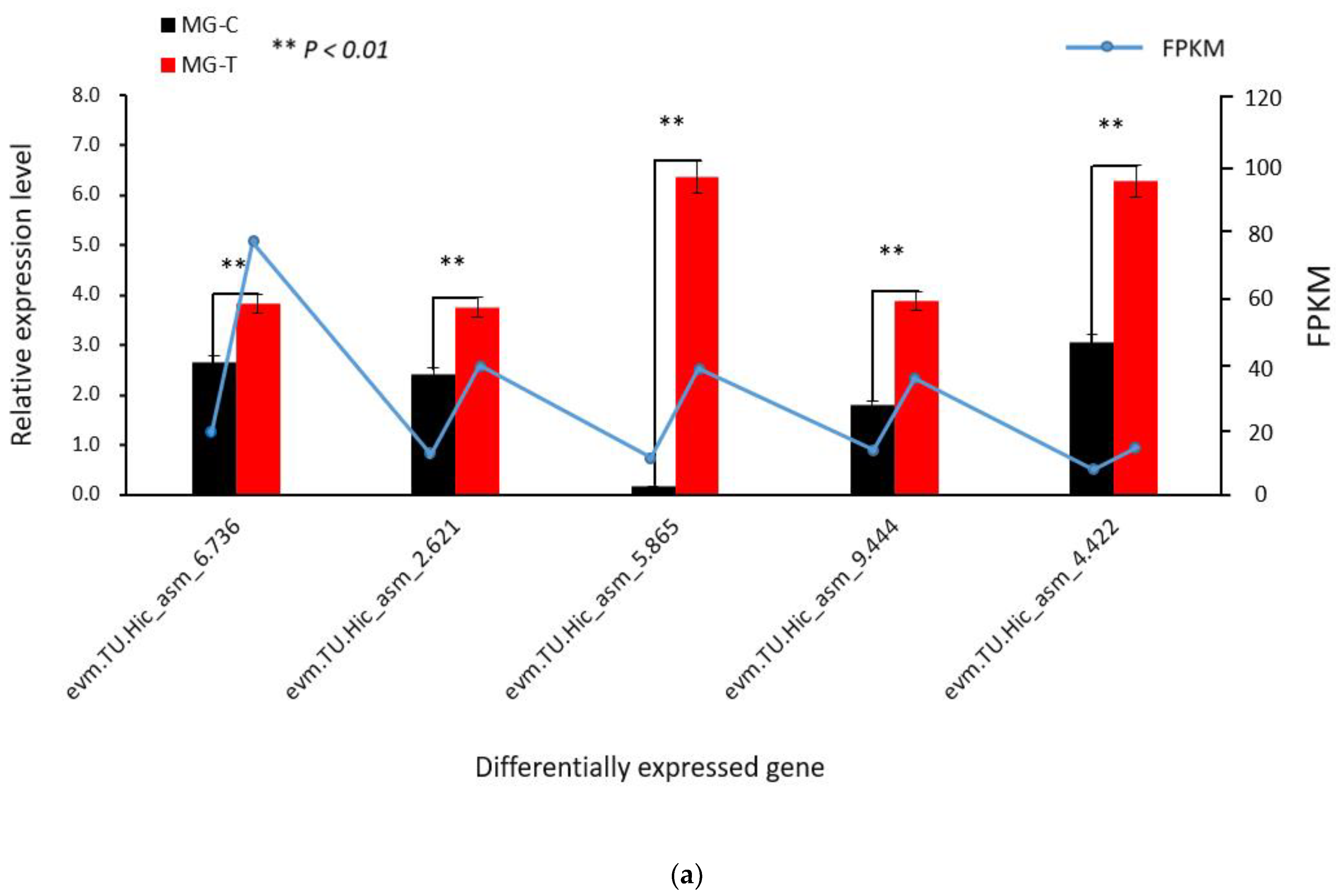
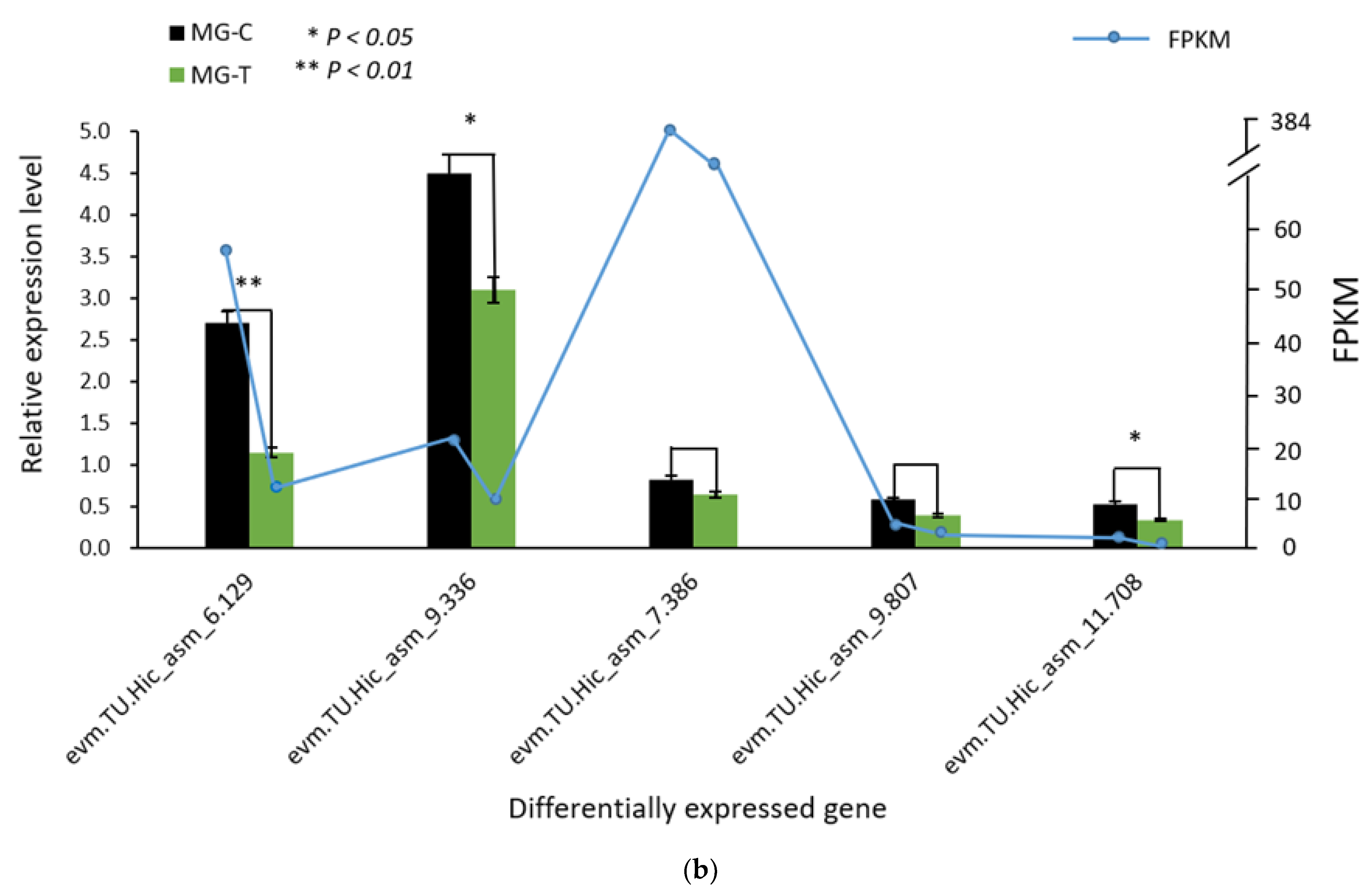
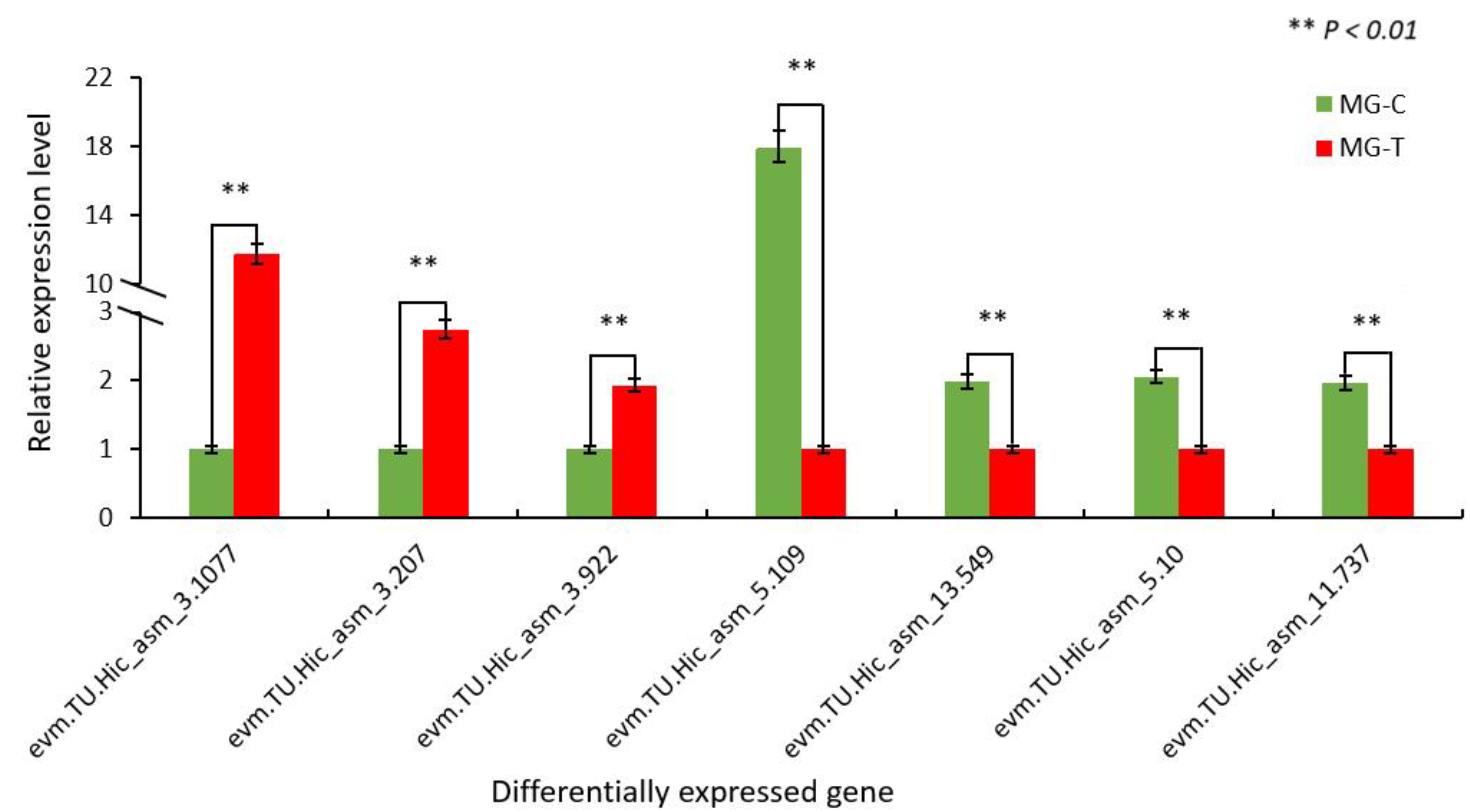
3.1.3. Verification of the Accuracy of Transcriptome Data by qRT-PCR
3.2. Proteomic Analysis of Midgut Samples
3.2.1. Protein Quantitative Analysis
3.2.2. Differentially Expressed Protein Statistics
3.2.3. Enrichment Analysis of DEPs
- GO enrichment analysis
- 2.
- KEGG Pathway Enrichment Analysis
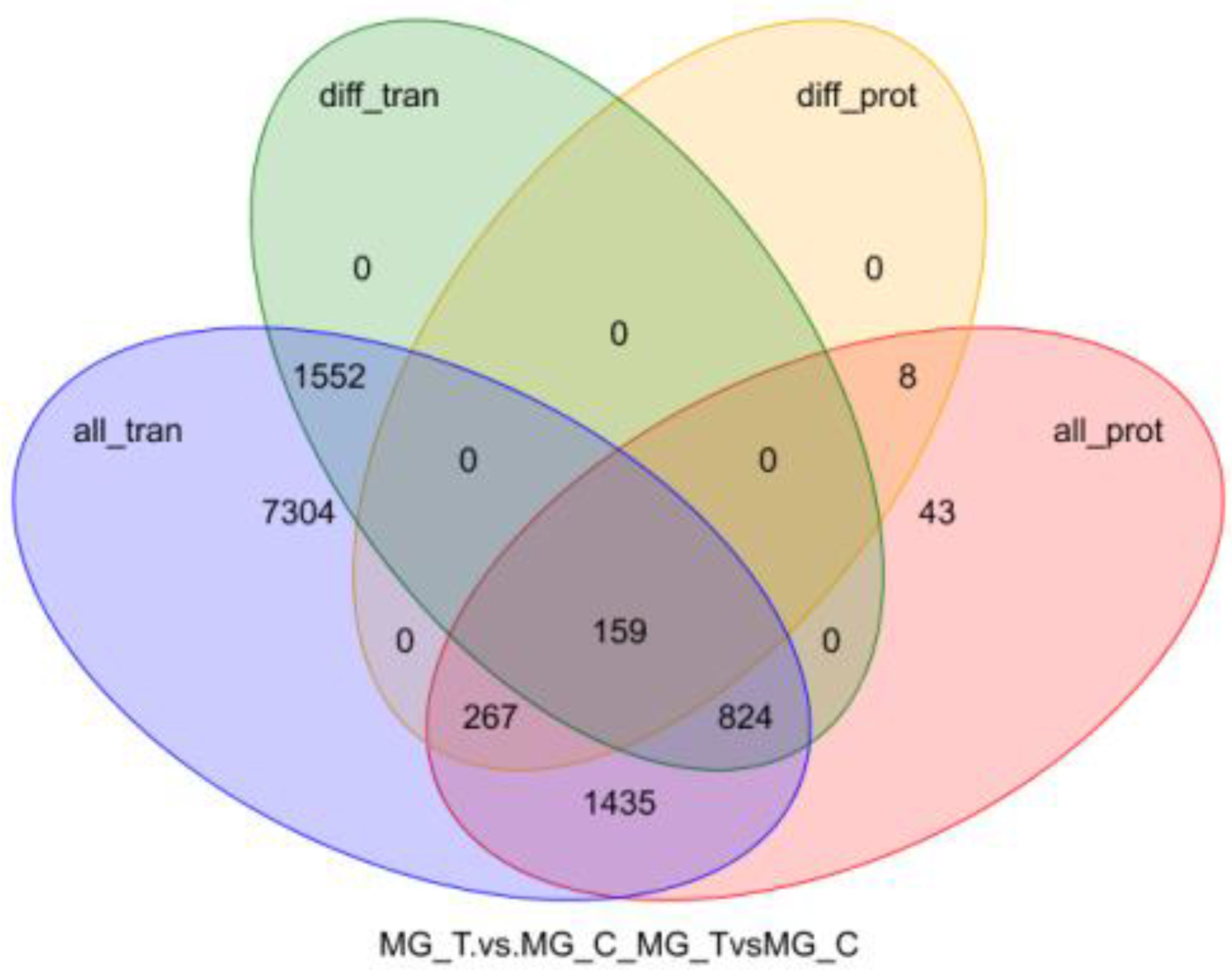
3.3. Association Analysis of Midgut Transcriptome and Proteome
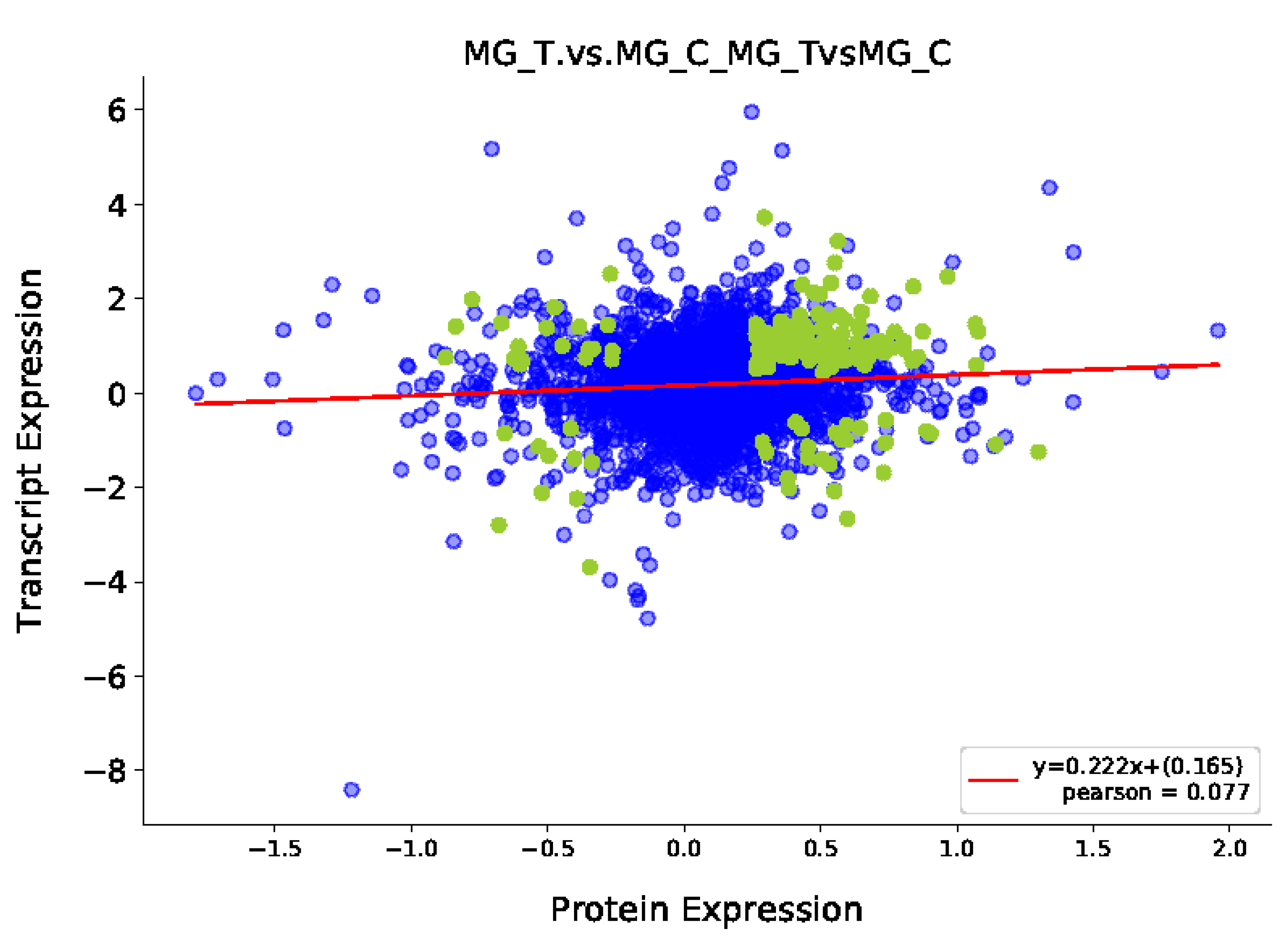
3.3.1. Transcriptome and Proteome Expression Regulation Analysis
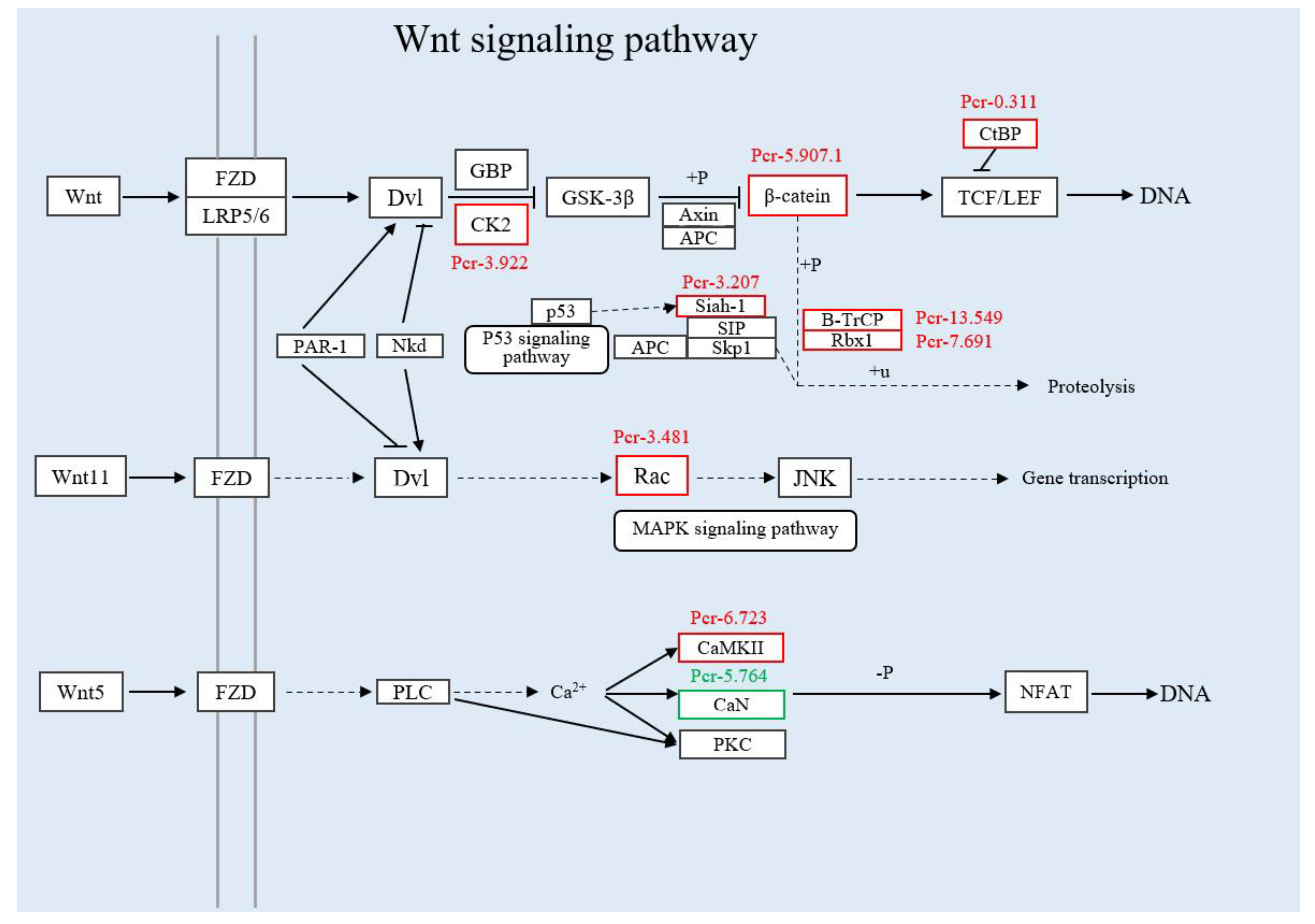
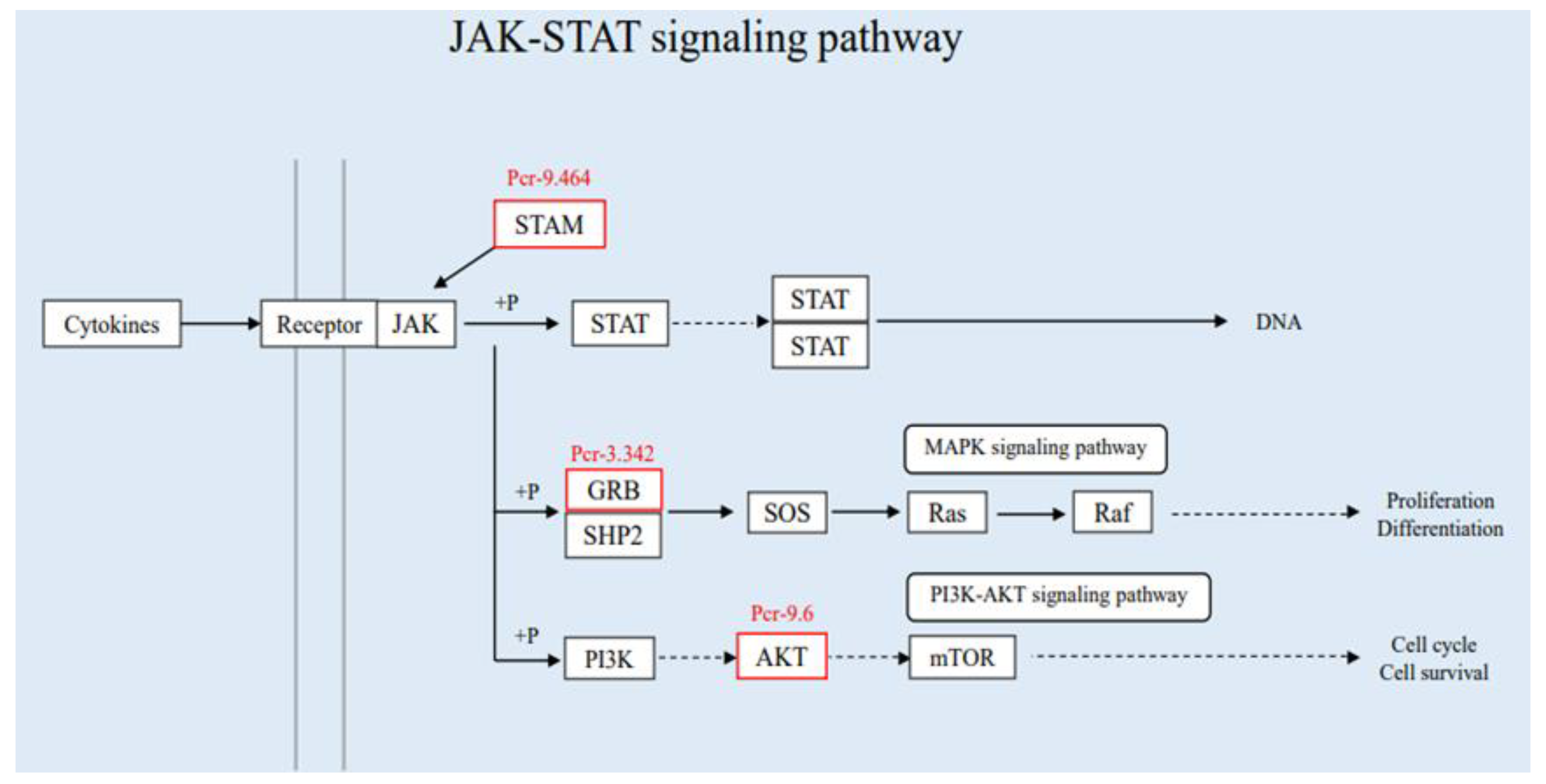
3.3.2. Joint Analysis of Transcriptome and Proteome Enrichment
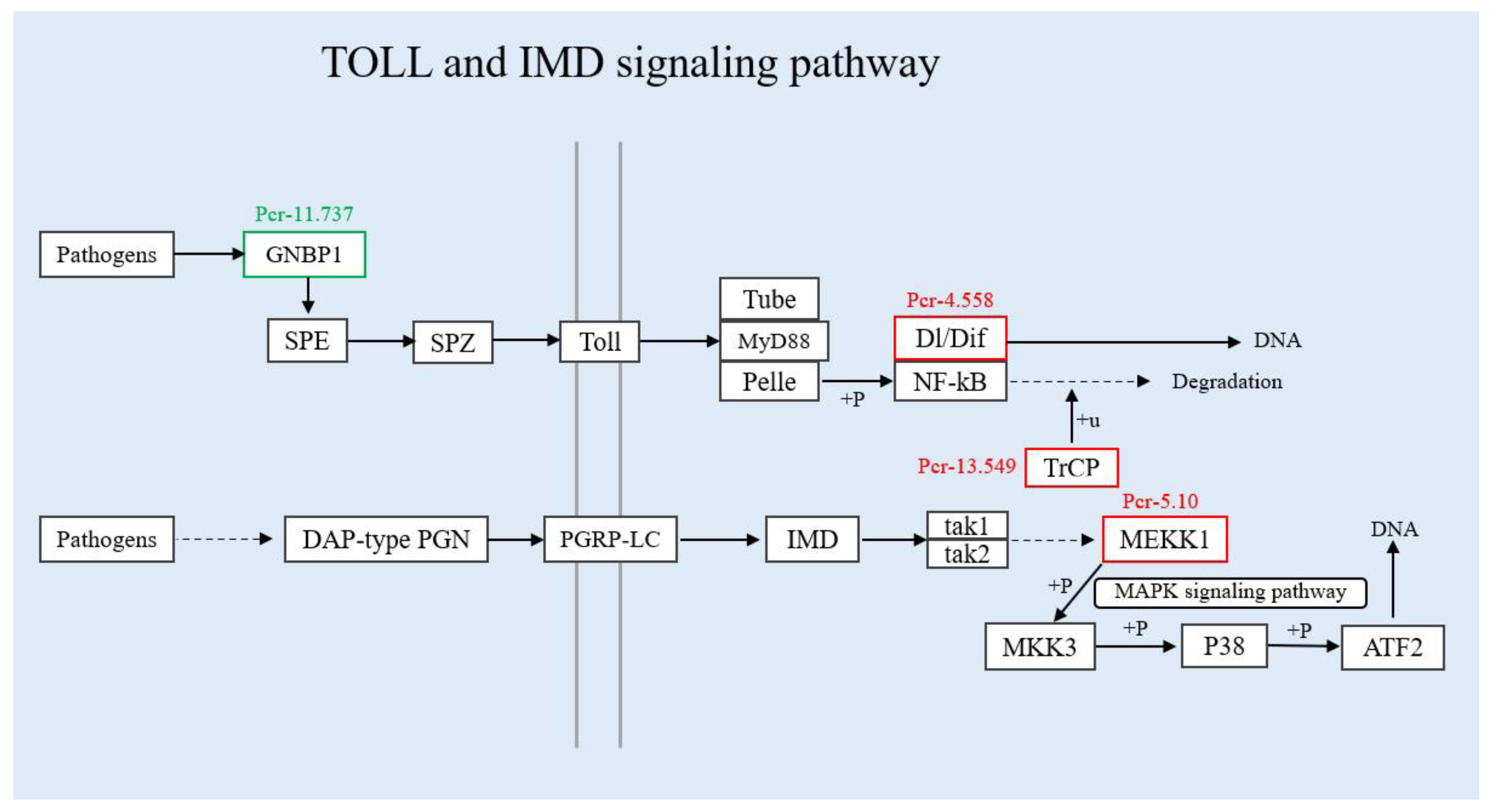
3.4. Verification of Differentially Expressed Genes in the Immune Pathway by qRT-PCR
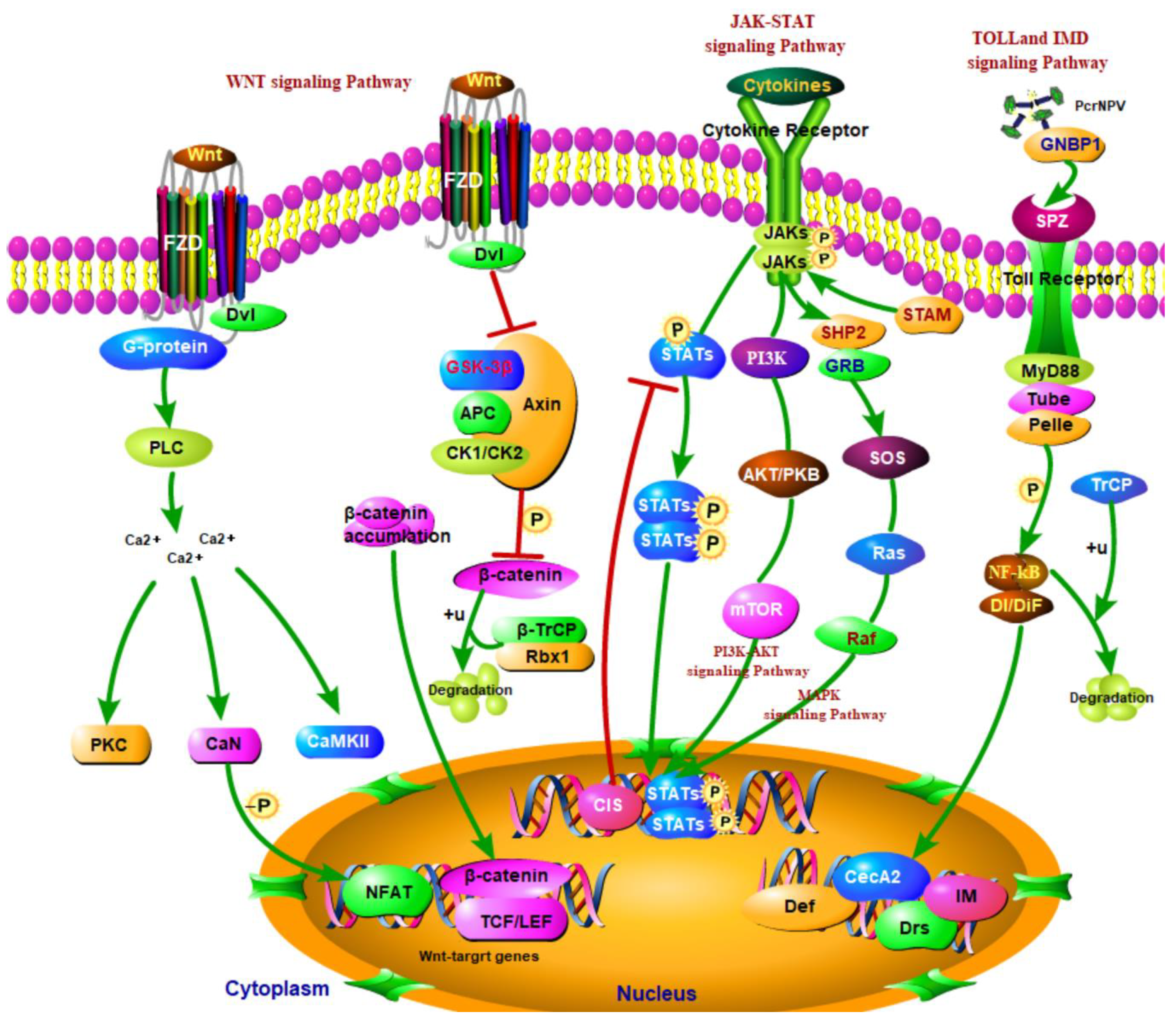
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhu, S.; Wang, W.; Wang, Y.; Yuan, M.; Yang, K. The baculovirus core gene ac83 is required for nucleocapsid assembly and per os infectivity of Autographa californica nucleopolyhedrovirus. J. Virol. 2013, 87, 10573–10586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.M.; Li, H. Research advances in baculovirus occlusion-derived virions. Bing Du Xue Bao 2016, 32, 93–100. [Google Scholar] [PubMed]
- Hillyer, J.F. Insect immunology and hematopoiesis. Dev. Comp. Immunol. 2016, 58, 102–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.S.; Wang, G.B.; Sun, Y.; Wei, L.; He, Y.Z.; Wang, F.C.; Jiang, Y.R.; Qin, L.; Joe, H.J. Transcriptome analysis of the midgut of the Chinese oak silkworm Antheraea pernyi infected with Antheraea pernyi nucleopolyhedrovirus. PLoS ONE 2016, 11, e0165959. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Ding, X.Y.; Chen, Q.Y.; Zhang, K.X.; Li, M.W. Bmapaf-1 is involved in the response against BmNPV Infection by the mitochondrial apoptosis pathway. Insects 2020, 11, 647. [Google Scholar] [CrossRef]
- Liu, Y.; Ai, J.W.; Tang, Y.; Xue, H.; He, X.J.; Zheng, Y. Expression analysis of antiviral genes related to BmNPV in silkworm(Bombyx mori) and screening of practical molecular markers. J. Agric. Biotechnol. 2019, 27, 2207–2215. [Google Scholar]
- Guo, H.; Xu, G.; Wang, B.B.; Xia, F.; Sun, Q.; Wang, Y.M.; Xie, E.Y.; Lu, Z.Y.; Jiang, L.; Xia, Q.Y. Phosphoenolpyruvate carboxykinase is involved in antiviral immunity against Bombyx mori nucleopolyhedrovirus. Dev. Comp. Immunol. 2019, 92, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Nishiyama, T.; Shigenobu, S.; Yamaguchi, K.; Suzuki, Y.; Shimada, T.; Katsuma, S.; Kiuchi, T. The genome sequence of Samia ricini, a new model species of lepidopteran insect. Mol. Ecol. Resour. 2021, 21, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Dewey, C.N.; Li, B. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Baren, M.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samarskiĭ, A.; Claverie, J.M. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139. [Google Scholar]
- Philip, J.; David, B.; Chang, H.Y.; Matthew, F.; Li, W.; Craig, M.A.; Hamish, M.W.; John, M.; Alex, M.; Gift, N. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar]
- Wei, H.D.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar]
- Livak, K.J.; Schmittgen, T. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔ Ct method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Kurasawa, J.H.; Park, A.; Sowers, C.R.; Halpin, R.A.; Ikeda, Y. Chemically defined, high-density insect cell-based expression system for scalable AAV vector production. Mol. Ther. Methods Clin. Dev. 2020, 19, 330–340. [Google Scholar] [CrossRef]
- Wang, X.Y.; Yu, H.Z.; Xu, J.P.; Zhang, S.Z.; Yu, D.; Liu, M.H.; Wang, L.L. Comparative subcellular proteomics analysis of susceptible and near-isogenic resistant Bombyx mori (Lepidoptera) larval midgut response to BmNPV infection. Sci. Rep. 2017, 7, 45690. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Guo, R.; Kumar, D.; Ma, H.; Liu, J.; Hu, X.; Cao, G.; Xue, R.; Gong, C. Identification, gene expression and immune function of the novel Bm-STAT gene in virus-infected Bombyx mori. Gene 2016, 577, 82–88. [Google Scholar] [CrossRef]
- Vanarsdall, A.; Mikhailov, V.; Rohrmann, G. Baculovirus DNA replication and processing. Curr. Drug Targets 2007, 8, 1096–1102. [Google Scholar] [CrossRef] [PubMed]
- Oomens, A.; Blissard, G.W. Requirement for GP64 to drive efficient budding of autographa californica multicapsid nucleopolyhedrovirus. Virology 1999, 254, 297–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, X.L.; Liu, T.H.; Wang, W.; Pan, C.X.; Pan, M.H. BmREEPa Is a novel gene that facilitates BmNPV entry into silkworm cells. PLoS ONE 2015, 10, e0144575. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.; Ankrah, R.; El-Tanani, S.; Loadman, P.M.; Pattterson, L.; Rudland, P.S.; El-Tanani, M. Wnt signaling as a therapeutic target in cancer and metastasis. In Introduction to Cancer Metastasis; Academic Press: Cambridge, MA, USA, 2017; pp. 375–394. [Google Scholar]
- Carlota, R.; Ainhoa, O.; Iolanda, L.; Beriat, M.; Jesus, E. Suppressor of cytokine signaling 1-derived peptide inhibits Janus kinase/signal transducers and activators of transcription pathway and improves inflammation and atherosclerosis in diabetic mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1953–1960. [Google Scholar]
- Boudny, V.; Kovarik, J. JAK/STAT signaling pathways and cancer Janus kinases/signal transducers and activators of transcription. Neoplasma 2002, 49, 349. [Google Scholar]
- Zhou, X.; Sun, Y.; Gao, J.; Sun, Y.; Liu, C.; Wang, L. Immune function of a Rab-related protein by modulating the JAK-STAT signaling pathway in the silkworm, Bombyx mori. Arch. Insect Biochem. Physiol. 2018, 97, 1. [Google Scholar] [CrossRef]
- Yang, Q.Q.; Tan, H.; Fu, Z.P.; Ma, Q.; Song, J.L. HSP90 inhibitor 17-AAG plays an important role in JAK3/STAT5 signaling pathways in HTLV-1 infection cell line HUT-102. Zhonghua Xue Ye Xue Za Zhi 2017, 38, 710–715. [Google Scholar]
- Rawlings, J.S. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef] [Green Version]
- Ren, F.; Wang, B.; Yue, T.; Yun, E.Y.; Ip, Y.T.; Jiang, J. Hippo signaling regulates drosophila intestine stem cell proliferation through multiple pathways. Proc. Natl. Acad. Sci. USA 2010, 107, 21064–21069. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Fan, X.; Li, W.X. Coactivation of STAT and Ras is required for germ cell proliferation and invasive migration in Drosophila. Dev. Cell 2003, 5, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.L.; Liu, J.B.; Lu, Y.H.; Gong, Y.C.; Zhu, M.; Chen, F.; Liang, Z.; Zhu, L.K.; Su, L.; Hu, X.L.; et al. Immune signaling pathways activated in response to different pathogenic micro-organisms in Bombyx mori. Mol. Immunol. 2015, 65, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Gregorio, D.E.; Spellman, P.T.; Tzou, P.; Rubin, G.M.; Lemaitre, B. The Toll and Imd pathways are the major regulators of the immune response in Drosophila. EMBO J. 2002, 21, 2568–2579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, M.; Qin, S.; Song, X.; Li, Y.; Jin, P. Evolutionary rate patterns of genes involved in the Drosophila Toll and Imd signaling pathway. BMC Evol. Biol. 2013, 13, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imler, J.L.; Zheng, L.B. Biology of Toll receptors: Lessons from insects and mammals. J. Leukoc. Biol. 2004, 75, 18–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanji, T.; Ip, Y.T. Regulators of the Toll and Imd pathways in the Drosophila innate immune response. Trends Immunol. 2005, 26, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Kaisho, T.; Akira, S. Toll-like receptors. Annu. Rev. Immunol. 2003, 21, 335–376. [Google Scholar] [CrossRef] [PubMed]
- Lamb, D.C.; Follmer, A.H.; Goldstone, J.V.; Nelson, D.R.; Warrilow, A.G.; Price, C.L.; True, M.Y.; Kelly, S.L.; Poulos, T.L.; Stegeman, J.J. On the occurrence of cytochrome P450 in viruses. Proc. Natl. Acad. Sci. USA 2019, 116, 12343–12352. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw-Reads | Clean-Reads | Clean-Bases | Error-Rate | Q20 (%) | Q30 (%) | GC-Pct |
|---|---|---|---|---|---|---|---|
| MG-C1 | 46,825,600 | 45,094,490 | 6.76G | 0.03% | 98.09 | 93.93 | 45.52% |
| MG-C2 | 46,803,912 | 44,600,114 | 6.69G | 0.03% | 98.08 | 93.93 | 45.61% |
| MG-C3 | 47,270,958 | 44,728,658 | 6.71G | 0.03% | 97.63 | 92.89 | 45.68% |
| MG-T1 | 47,180,212 | 44,402,340 | 6.66G | 0.02% | 98.13 | 94.12 | 45.17% |
| MG-T2 | 48,422,268 | 46,367,536 | 6.96G | 0.03% | 96.49 | 90.81 | 44.96% |
| MG-T3 | 47,769,512 | 44,863,038 | 6.73G | 0.02% | 98.26 | 94.29 | 43.20% |
| DEGs | All | Up | Down | Threshold | ||
|---|---|---|---|---|---|---|
| MG-T vs. MG-C | 2535 | 1264 | 1271 | DESeq2 | padj ≤ 0.05 | |log2(FC)| ≥ 1.5 |
| GO Classification | GO Function | Number of Unigenes | Up-Regulated | Down-Regulated |
|---|---|---|---|---|
| Biological Process | translation | 78 | 77 | 1 |
| peptide metabolic process | 80 | 79 | 1 | |
| peptide biosynthetic process | 78 | 77 | 1 | |
| amide biosynthetic process | 78 | 77 | 1 | |
| organonitrogen compound biosynthetic process | 109 | 103 | 6 | |
| macromolecule biosynthetic process | 168 | 118 | 50 | |
| gene expression | 170 | 119 | 51 | |
| cellular nitrogen compound biosynthetic process | 165 | 115 | 50 | |
| cellular amide metabolic process | 81 | 79 | 2 | |
| cellular macromolecule biosynthetic process | 167 | 118 | 47 | |
| Cellular Component | cytoplasmic part | 114 | 106 | 8 |
| ribonucleoprotein complex | 68 | 68 | 0 | |
| ribosome | 58 | 58 | 0 | |
| cytoplasm | 127 | 115 | 12 | |
| protein-containing complex | 144 | 112 | 32 | |
| non-membrane-bounded organelle | 77 | 63 | 14 | |
| intracellular non-membrane-bounded organelle | 77 | 63 | 14 | |
| mitochondrion | 29 | 27 | 2 | |
| mitochondrial part | 21 | 19 | 2 | |
| peptidase complex | 15 | 14 | 1 | |
| Molecular Function | structural constituent of ribosome | 55 | 55 | 0 |
| structural molecule activity | 63 | 59 | 4 | |
| ATP binding | 150 | 69 | 81 | |
| adenyl ribonucleotide binding | 150 | 69 | 81 | |
| adenyl nucleotide binding | 150 | 69 | 81 | |
| cytoskeletal protein binding | 26 | 6 | 20 | |
| threonine-type endopeptidase activity | 11 | 11 | 0 | |
| threonine-type peptidase activity | 11 | 11 | 0 | |
| RNA-binding | 46 | 38 | 8 | |
| microtubule binding | 13 | 4 | 9 |
| KEGG ID | Pathway | No. of Unigenes | Up-Regulated | Down-Regulated | Padj |
|---|---|---|---|---|---|
| bmor03010 | Ribosome | 72 | 72 | 0 | 3.67 × 10−13 |
| bmor03008 | Ribosome biogenesis in eukaryotes | 39 | 36 | 3 | 1.10 × 10−6 |
| bmor00190 | Oxidative phosphorylation | 49 | 49 | 0 | 1.45 × 10−5 |
| bmor03060 | Protein export | 15 | 14 | 1 | 0.000124708 |
| bmor03050 | Proteasome | 22 | 22 | 0 | 0.002445682 |
| Samples | Number of Co-Identified Proteins | Regulated Type | FC > 1.2 | FC > 1.3 | FC > 1.5 | FC > 2.0 |
|---|---|---|---|---|---|---|
| MG-T vs. MG-C | 2971 | up-regulated | 360 | 264 | 118 | 20 |
| down-regulated | 102 | 66 | 27 | 3 |
| GO Classification | GO Term | x/n | Up | Down | p-Value | GO ID | |
|---|---|---|---|---|---|---|---|
| Biological Process | cellular macromolecule metabolic process | 67/313 | 21.41% | 60 | 7 | 0.030463 | GO:0044260 |
| cellular nitrogen compound metabolic process | 50/313 | 15.97% | 41 | 9 | 0.045171 | GO:0034641 | |
| nucleobase-containing compound metabolic process | 31/313 | 9.90% | 27 | 4 | 0.046252 | GO:0006139 | |
| ribonucleoprotein complex biogenesis | 6/313 | 1.92% | 6 | 0 | 0.048092 | GO:0022613 | |
| Cellular Component | intracellular membrane-bounded organelle | 43/313 | 13.74% | 36 | 7 | 0.002168 | GO:0043231 |
| nucleolus | 23/313 | 7.35% | 21 | 2 | 0.009377 | GO:0005634 | |
| organelle | 59/313 | 18.85% | 48 | 11 | 0.025455 | GO:0043226 | |
| intracellular organelle | 58/313 | 18.53% | 48 | 10 | 0.036687 | GO:0043229 | |
| Molecular Function | nucleic acid binding | 48/313 | 15.34% | 42 | 6 | 0.010857 | GO:0003676 |
| glucosidase activity | 2/313 | 0.64% | 2 | 0 | 0.021454 | GO:0015926 | |
| Ran GTPase binding | 3/313 | 0.96% | 3 | 0 | 0.024863 | GO:0008536 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, G.; Zhang, B.; Zhang, H.; Xu, A.; Qian, H. Integration of Transcriptomic and Proteomic Analyses Reveals New Insights into the Regulation of Immune Pathways in Midgut of Samia ricini upon SariNPV Infection. Insects 2022, 13, 294. https://doi.org/10.3390/insects13030294
Li G, Zhang B, Zhang H, Xu A, Qian H. Integration of Transcriptomic and Proteomic Analyses Reveals New Insights into the Regulation of Immune Pathways in Midgut of Samia ricini upon SariNPV Infection. Insects. 2022; 13(3):294. https://doi.org/10.3390/insects13030294
Chicago/Turabian StyleLi, Gang, Benzheng Zhang, Huan Zhang, Anying Xu, and Heying Qian. 2022. "Integration of Transcriptomic and Proteomic Analyses Reveals New Insights into the Regulation of Immune Pathways in Midgut of Samia ricini upon SariNPV Infection" Insects 13, no. 3: 294. https://doi.org/10.3390/insects13030294