
Comparative Transcriptome Investigation of Nosema ceranae Infecting Eastern Honey Bee Workers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Fungal Spore and Honey Bee
2.2. Microscopic Observation and PCR Validation of N. ceranae Spores
2.3. Preparation and Detection of Paraffin Section of Honey Bee Midgut Tissue
2.4. Transcriptome Data Source
2.5. Identification and Analysis of DEGs in N. ceranae
2.6. Investigation of Virulence Factor-Associated DEGs
2.7. RT-qPCR Validation of DEGs
2.8. Statistical Analysis
3. Results
3.1. Verification of Infection of A. c. cerana Worker by N. ceranae
3.2. Differential Gene Expression Profile of N. ceranae Infecting A. c. cerana Workers
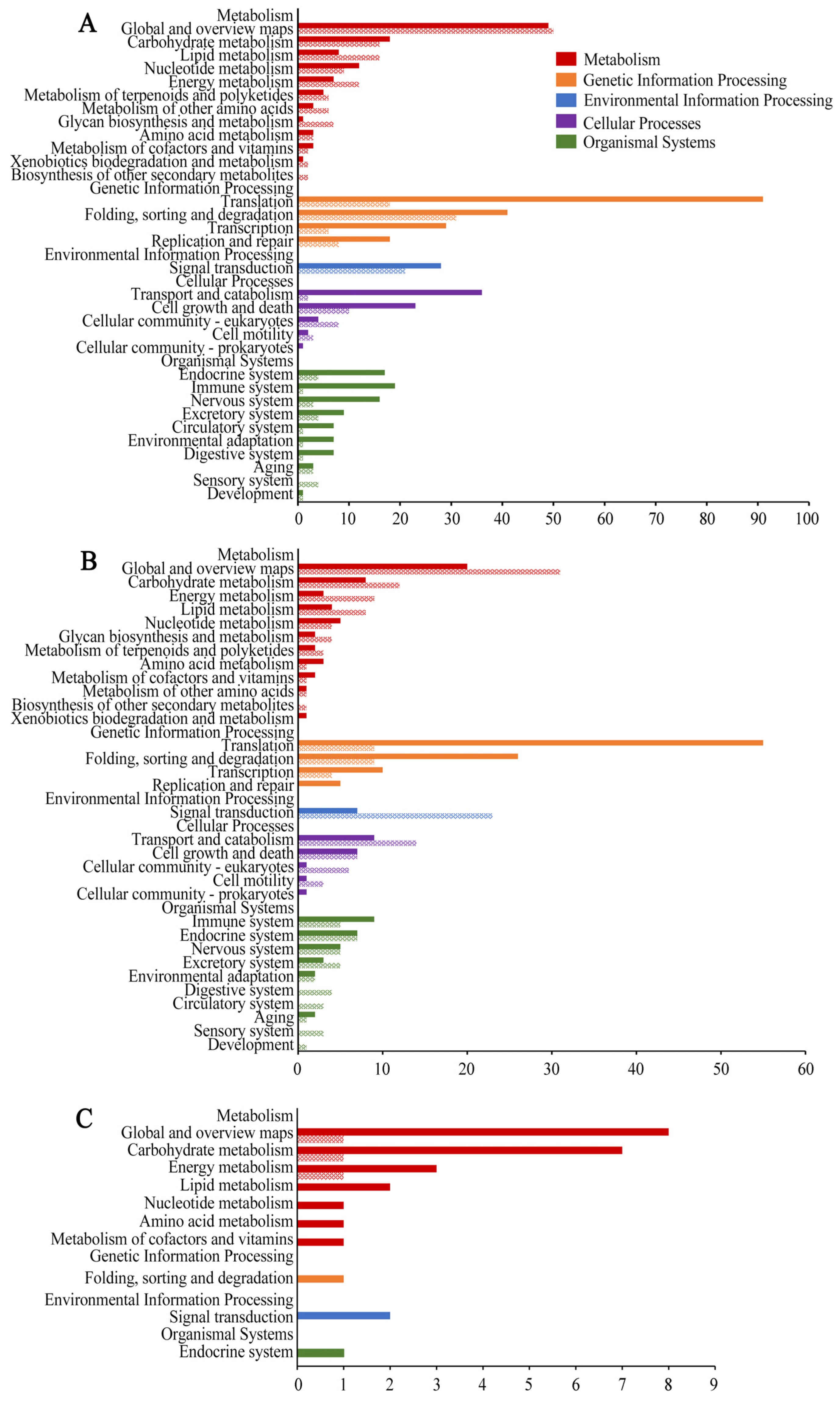
3.3. Function and Pathway Annotation of DEGs in N. ceranae Infesting A. c. cerana Workers
3.4. Virulence Factor-Associated DEGs in N. ceranae Invading A. c. cerana Workers
3.5. Verification of DEGs via RT-qPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fries, I.; Feng, F.; Silva, A.D.; Slemenda, S.B.; Pieniazek, N.J. Nosema ceranae, n. sp. (Microspora, Nosematidae), morphological and molecular characterization of a microsporidian parasite of the Asian honey bee Apis cerana, (Hymenoptera, Apidae). Eur. J. Protistol. 1996, 32, 356–365. [Google Scholar] [CrossRef]
- Martín-Hernández, R.; Meana, A.; Prieto, L.; Salvador, A.M.; Garrido-Bailón, E.; Garrido-Bailón, E.; Higes, M. Outcome of colonization of Apis mellifera by Nosema ceranae. Appl. Environ. Microbiol. 2007, 73, 6331–6338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayack, C.; Naug, D. Energetic stress in the honey bee Apis mellifera from Nosema ceranae infection. J. Invertebr. Pathol. 2009, 100, 185–188. [Google Scholar] [CrossRef]
- Zheng, S.B.; He, J.F.; Li, Z.G.; Gao, Z.S.; Yu, T.T.; Xu, W.J.; Su, S.K. Effects of Nosema ceranae on the expression of immune gene and hemolymph sugar levels of Apis cerana bees. Entomol. Knowl. 2017, 54, 392–399. [Google Scholar]
- Kurze, C.; Le, C.Y.; Dussaubat, C.; Erler, S.; Kryger, P.; Lewkowski, O.; Müller, T.; Widder, M.; Moritz, R.F. Nosema tolerant honey bees (Apis mellifera) escape parasitic manipulation of apoptosis. PLoS ONE 2015, 10, e0140174. [Google Scholar] [CrossRef]
- Fries, I. Nosema ceranae in European honey bees (Apis mellifera). J. Invertebr. Pathol. 2010, 103, S73–S79. [Google Scholar] [CrossRef]
- Lach, L.; Kratz, M.; Boris, B. Parasitized honey bees are less likely to forage and carry less pollen. J. Invertebr. Pathol. 2015, 130, 64–71. [Google Scholar] [CrossRef]
- Dussaubat, C.; Maisonnasse, A.; Crauser, D.; Beslay, D.; Costagliola, G.; Soubeyrand, S.; Kretzchmar, A.; Conte, Y.L. Flight behavior and pheromone changes associated to Nosema ceranae infection of honey bee workers (Apis mellifera) in field conditions. J. Invertebr. Pathol. 2013, 113, 42–51. [Google Scholar] [CrossRef]
- Higes, M.; Martín-Hernández, R.; Garrido-Bailón, E.; González-Porto, A.V.; García-Palencia, P.; Meana, A.; Nozal, M.J.D.; Mayo, R.; Bernal, J.L. Honey bee colony collapse due to Nosema ceranae in professional apiaries. Environ. Microbiol. Rep. 2009, 1, 110–113. [Google Scholar] [CrossRef]
- Bromenshenk, J.J.; Henderson, C.B.; Wick, C.H.; Stanford, M.F.; Zulich, A.W.; Jabbour, R.E.; Deshpande, S.V.; McCubbin, P.E.; Seccomb, R.A.; Welch, P.M.; et al. Iridovirus and microsporidian linked to honey bee colony decline. PLoS ONE 2010, 5, e13181. [Google Scholar] [CrossRef]
- Chen, Y.P.; Evans, J.D.; Charles, M.; Robin, G.; Michael, Z.; Gundensen-Rindal, D.; Pettis, J.S. Morphological, molecular, and phylogenetic characterization of Nosema ceranae, a microsporidian parasite isolated from the European honey bee, Apis mellifera. J. Eukaryot. Microbiol. 2009, 56, 142–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.F.; Solter, L.F. Comparative development and tissue tropism of Nosema apis and Nosema ceranae. J. Invertebr. Pathol. 2013, 113, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Badaoui, B.; Fougeroux, A.; Petit, F.; Anselom, A.; Gorni, C.; Cucurachi, M.; Cersini, A.; Granato, A.; Cardeti, G.; Formato, G.; et al. RNA-sequence analysis of gene expression from honey bees (Apis mellifera) infected with Nosema ceranae. PLoS ONE 2017, 12, e0173438. [Google Scholar] [CrossRef]
- Dussaubat, C.; Brunet, J.; Higes, M.; Colbourne, J.K.; Lopez, J.; Choi, J.H.; Martín-Hernández, R.; Botías, C.; Cousin, M.; McDonnell, C.; et al. Gut pathology and responses to the microsporidium Nosema ceranae in the honey bee Apis mellifera. PLoS ONE 2012, 7, e37017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panek, J.; Paris, L.; Roriz, D.; Mone, A.; Dubuffet, A.; Delbac, F.; Diogon, M.; Alaoui, H. Impact of the microsporidian Nosema ceranae on the gut epithelium renewal of the honey bee, Apis mellifera. J. Invertebr. Pathol. 2018, 159, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Zhou, D.; Chen, H.; Xiong, C.; Zheng, Y.; Chen, D.; Guo, R. MicroRNA dataset of normal and Nosema ceranae-infected midguts of Apis cerana cerana workers. Data Brief. 2019, 26, 104518. [Google Scholar] [CrossRef]
- Chen, D.; Chen, H.; Du, Y.; Zhou, D.; Geng, S.; Wang, H.; Wan, J.; Xiong, C.; Zheng, Y.; Guo, R. Genome-wide identification of long non-coding RNAs and their regulatory networks involved in Apis mellifera ligustica response to Nosema ceranae infection. Insects 2019, 10, 245. [Google Scholar] [CrossRef] [Green Version]
- Guo, R.; Chen, D.; Xiong, C.; Zheng, Y.; Hou, C.; Du, Y.; Geng, S.; Wang, H.; Zhou, D.; Guo, Y. Genome-wide identification of circular RNAs in fungal parasite Nosema ceranae. Curr. Microbiol. 2018, 29, 1655–1660. [Google Scholar] [CrossRef]
- Fu, Z.; Zhou, D.; Chen, H.; Geng, S.; Chen, D.; Zhen, Y.; Xiong, C.; Xu, G.; Zhang, X.; Guo, R. Analysis of highly expressed genes in Apis cerana cerana workers midguts responding to Nosema ceranae stress. J. Sichuan Univ. (Nat. Sci. Ed.) 2020, 57, 191–198. [Google Scholar] [CrossRef]
- Xiong, C.; Chen, H.; Geng, S.; Zhou, N.; Zhou, D.; Zhu, Z.; Chen, D.; Zheng, Y.; Xu, G.; Zhang, X.; et al. Expression profile of high-expressing genes and its potential role during Apis cerana cerana infected by Nosema ceranae. J. Sichuan Univ. (Nat. Sci. Ed.) 2020, 57, 596–604. [Google Scholar] [CrossRef]
- Xing, W.; Zhou, D.; Long, Q.; Sun, M.; Guo, R.; Wang, L. Immune Response of Eastern Honey bee Worker to Nosema ceranae Infection Revealed by Transcriptomic Investigation. Insects 2021, 12, 728. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Du, Y.; Chen, H.; Fan, Y.; Fan, X.; Zhu, Z.; Wang, J.; Xiong, C.; Zheng, Y.; Hou, C.; et al. Comparative identification of microRNAs in Apis cerana cerana workers’ midguts in response to Nosema ceranae invasion. Insects 2019, 10, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Evans, J.D.; Smith, I.B.; Pettis, J.S. Nosema ceranae is a long-present and wide-spread microsporidian infection of the European honey bee (Apis mellifera) in the United States. J. Invertebr. Pathol. 2008, 97, 186–188. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Carthy, D.M.; Robinson, M.; Robinson, M.; Smyth, G. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Lin, F.; Hongkun, Z.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2005, 2, 293–297. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef]
- Du, Y.; Fan, X.; Jiang, H.; Wang, J.; Feng, R.; Zhang, W.; Yu, K.; Long, Q.; Cai, Z.; Xiong, C.; et al. MicroRNA-mediated cross-kingdom regulation of Apis mellifera ligustica worker to Nosema ceranae. Sci. Agric. Sin. 2021, 54, 1805–1820. [Google Scholar] [CrossRef]
- Fan, X.; Du, Y.; Zhang, W.; Wang, J.; Jiang, H.; Fan, Y.; Feng, R.; Wang, J.; Zhou, Z.; Xiong, C.; et al. Omics analysis of Nosema ceranae miRNAs involved in gene expression regulation in the midgut of Apis mellifera ligustica workers and their regulatory networks. Acta Entomol. Sin. 2021, 64, 187–204. [Google Scholar] [CrossRef]
- Geng, S.; Zhou, D.; Fan, X.; Jiang, H.; Zhu, Z.; Wang, J.; Fan, Y.; Wang, X.; Xiong, C.; Zheng, Y.; et al. Transcriptomic analysis reveals the molecular mechanism underlying Nosema ceranae infection of Apis mellifera ligustica. Acta Entomol. Sin. 2020, 63, 294–308. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Pérez, P.; Cansado, J. Cell integrity signaling and response to stress in fission yeast. Curr. Protein Pept. Sci. 2010, 11, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, M.; Cai, S.; He, X.; Shao, Y.; Lu, X. Ricin-B-lectin enhances microsporidia Nosema bombycis infection in BmN cells from silkworm Bombyx mori. Acta Biochim. Biophys. Sin. 2016, 48, 1020–1057. [Google Scholar] [CrossRef] [Green Version]
- Qi, X. Studies on the Functional Genome of Nosema Bombycis-Localization and Functional Analysis on Ricin B-Lectin NbRBL3 of Nosema bombycis; Chongqing Southwest University: Chongqing, China, 2014; pp. 1–88. [Google Scholar]
- Long, M.; Tan, Y.; Yu, B.; Pan, G.; Zhou, Z. Expression of Nosema bombycis polar tube protein 1 in lepidopteran sf9 cells and its effect on microsporidian proliferation. J. Invertebr. Pathol. 2020, 172, 107350. [Google Scholar] [CrossRef]
- Wu, Z.; Li, Y.; Pan, J.; Zhou, Z.; Xiang, Z. SWP25, a novel protein associated with the Nosema bombycis endospore. J. Eukaryot. Microbiol. 2009, 56, 113–118. [Google Scholar] [CrossRef]
- Yang, D.; Pan, L.; Peng, P.; Dang, X.; Li, C.; Li, T.; Long, M.; Chen, J.; Wu, Y.; Du, H.; et al. Interaction between SWP9 and polar tube proteins of the microsporidian Nosema bombycis and function of SWP9 as a scaffolding protein contribute to polar tube tethering to the spore wall. Infect. Immun. 2017, 85, e00872-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Pan, L.; Chen, Z.; Du, H.; Luo, B.; Luo, J.; Pan, G. The roles of microsporidia spore wall proteins in the spore wall formation and polar tube anchorage to spore wall during development and infection processes. Exp. Parasitol. 2018, 187, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Long, M.; Tan, Y.; Liu, K.; Wu, Y.; Lü, Q.; Pan, G.; Zhou, Z. Expression of Polar Tube Protein 1 (NbPTP1) from Nosema bombycis in Drosophila S2 cell lines and its glycosylation. Chin. J. Biotechnol. 2018, 34, 1460–1468. [Google Scholar] [CrossRef]
- Huang, Q.; Chen, Y.; Wang, R.; Cheng, S.; Evans, J. Host-parasite interactions and purifying selection in a microsporidian parasite of honey bees. PLoS ONE 2016, 11, e0147549. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Peng, X.; Li, X.; Kye, I.B.; Shang, R.; Qi, J.; Zhang, Y.; Xu, L.; Sheng, Z. Cloning, sequence structure analysis and prokaryotic expression of Nosema bombycis hexokinase gene. Acta Sericologica Sin. 2018, 44, 70–76. [Google Scholar] [CrossRef]
- Zhang, Z.; Qi, J.; Shang, R.; Chen, H.; Zhang, Y.; Sheng, Z. Cloning and expression characteristics of pyruvate kinase gene of Nosema bombycis. Acta Sericologica Sin. 2019, 45, 212–217. [Google Scholar] [CrossRef]
- Yao, Q.; Yang, Y.; Cui, L.; Su, H.; Li, R.; Lu, G. Elementary analysis of 6-phosphofructokinase gene of Xanthomonas campestris. Genom. Appl. Biol. 2010, 29, 1047–1054. [Google Scholar] [CrossRef]
- Dermauw, W.; Leeuwen, T.V. The ABC gene family in arthropods comparative genomics and role in insecticide transport and resistance. Insect Biochem. Mol. Biol. 2014, 45, 89–110. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Y.; Wang, J.; Yu, K.; Zhang, W.; Cai, Z.; Sun, M.; Hu, Y.; Zhao, X.; Xiong, C.; Niu, Q.; et al. Comparative Transcriptome Investigation of Nosema ceranae Infecting Eastern Honey Bee Workers. Insects 2022, 13, 241. https://doi.org/10.3390/insects13030241
Fan Y, Wang J, Yu K, Zhang W, Cai Z, Sun M, Hu Y, Zhao X, Xiong C, Niu Q, et al. Comparative Transcriptome Investigation of Nosema ceranae Infecting Eastern Honey Bee Workers. Insects. 2022; 13(3):241. https://doi.org/10.3390/insects13030241
Chicago/Turabian StyleFan, Yuanchan, Jie Wang, Kejun Yu, Wende Zhang, Zongbing Cai, Minghui Sun, Ying Hu, Xiao Zhao, Cuiling Xiong, Qingsheng Niu, and et al. 2022. "Comparative Transcriptome Investigation of Nosema ceranae Infecting Eastern Honey Bee Workers" Insects 13, no. 3: 241. https://doi.org/10.3390/insects13030241