Invasion History of Sirex noctilio Based on COI Sequence: The First Six Years in China


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Selection
2.2. DNA Sequence Analysis
2.3. Statistical Analysis
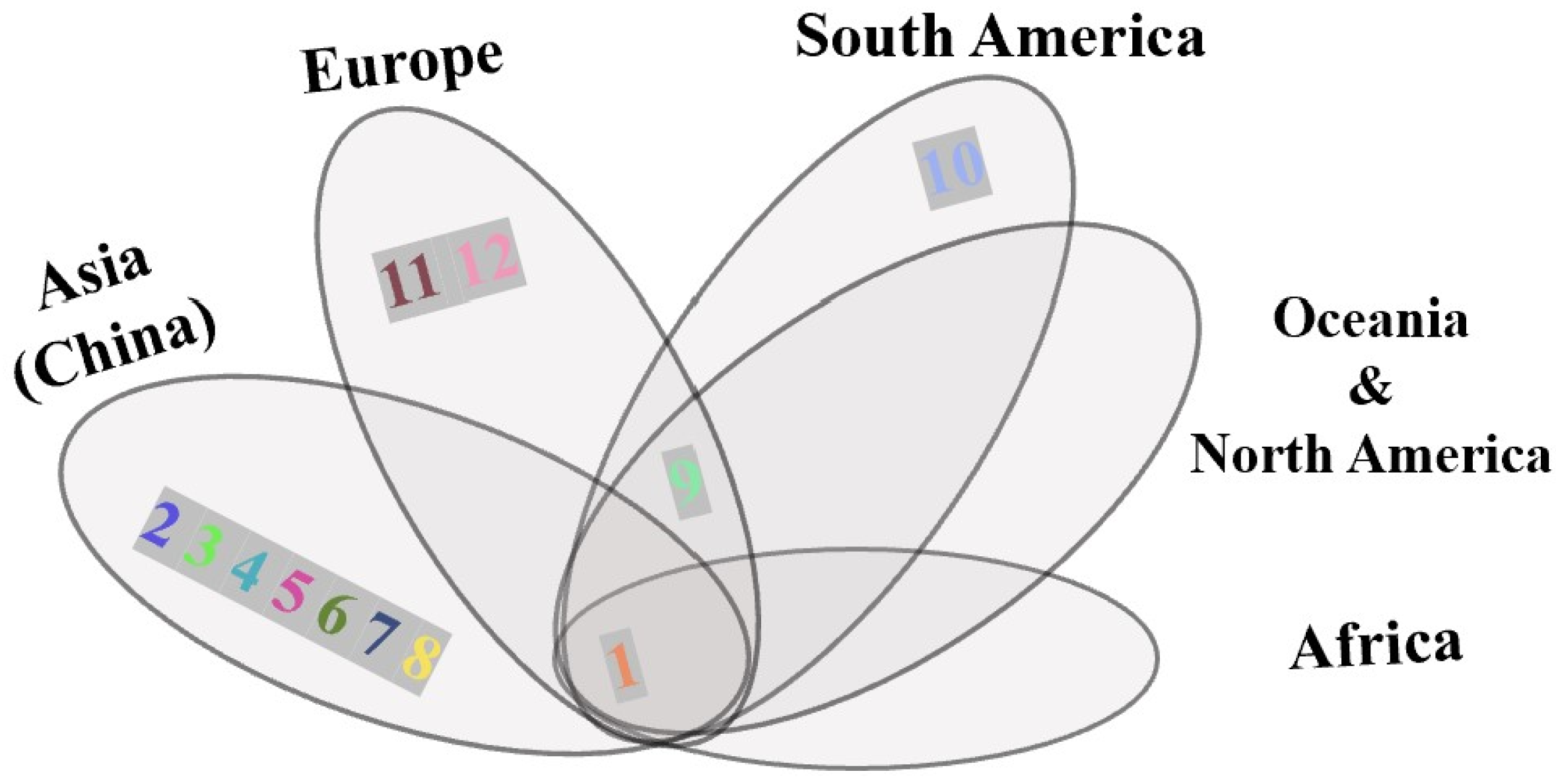
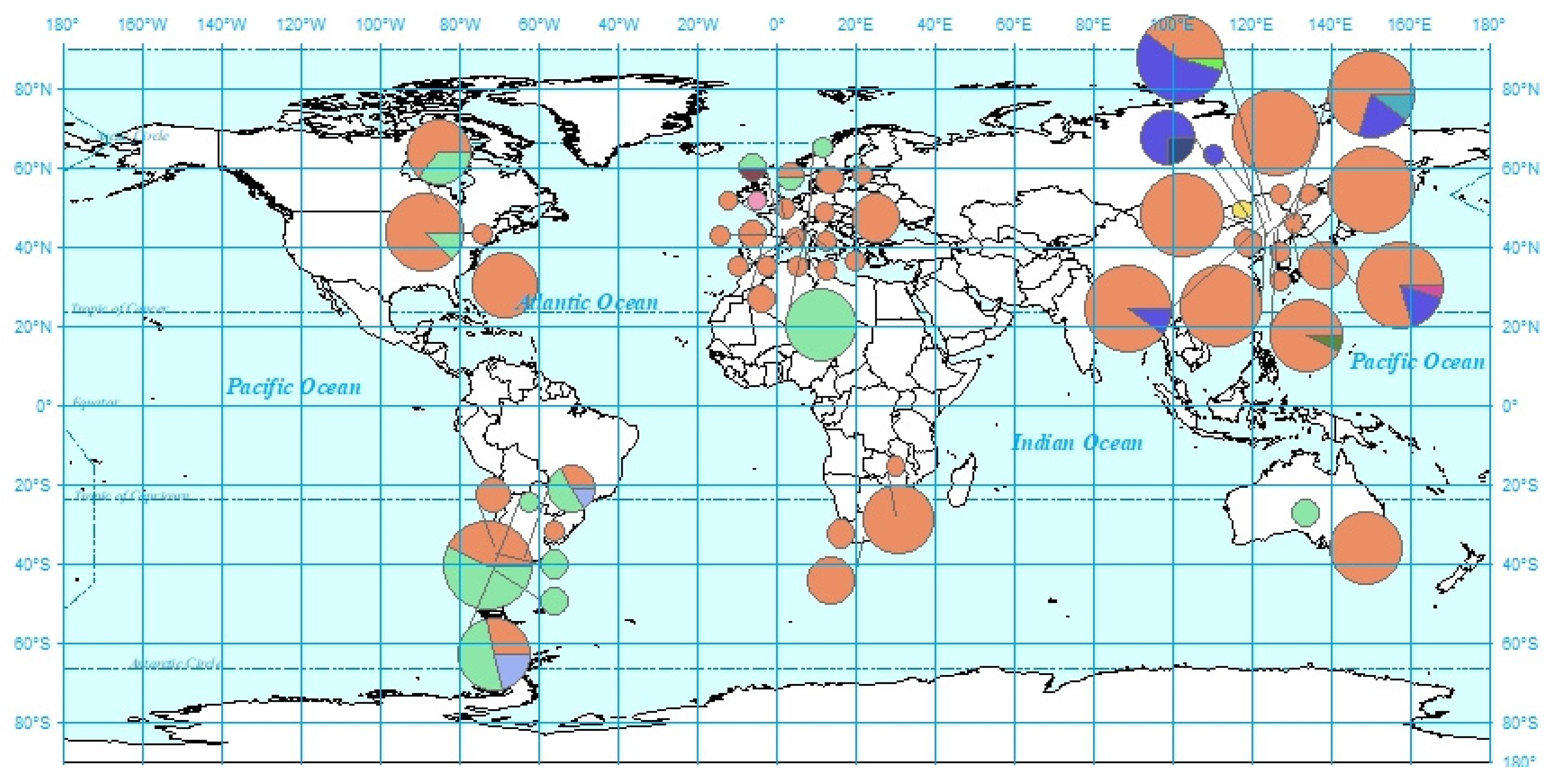
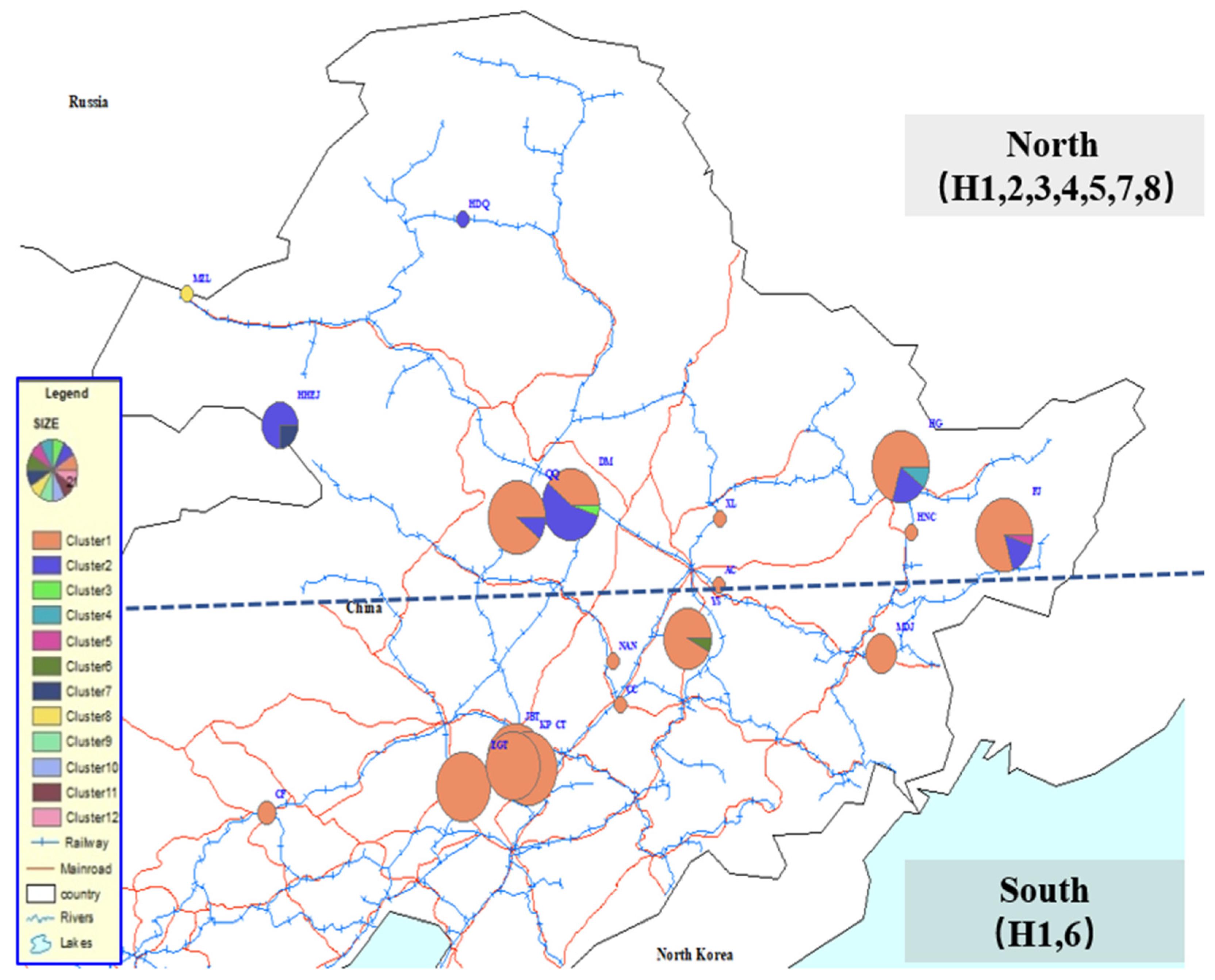
3. Results
4. Discussion
4.1. Host Unity of S. Noctilio in China
4.2. Patterns of Multiple Invasion and Spread in China
4.3. Focus for Further Research
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hulme, P.E. Trade, transport and trouble: Managing invasive species pathways in an era of globalization. J. Appl. Ecol. 2009, 46, 10–18. [Google Scholar] [CrossRef]
- Seebens, H.; Blackburn, T.M.; Dyer, E.E.; Genovesi, P.; Hulme, P.E.; Jeschke, J.M.; Pagad, S.; Pyšek, P.; Winter, M.; Arianoutsou, M. No saturation in the accumulation of alien species worldwide. Nat. Commun. 2017, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Roques, A.; Auger-Rozenberg, M.-A.; Blackburn, T.M.; Garnas, J.; Pyšek, P.; Rabitsch, W.; Richardson, D.M.; Wingfield, M.J.; Liebhold, A.M.; Duncan, R.P. Temporal and interspecific variation in rates of spread for insect species invading Europe during the last 200 years. Biol. Invasions 2016, 18, 907–920. [Google Scholar] [CrossRef]
- Simberloff, D. Nonindigenous Species: A Global Threat to Biodiversity and Stability; Nature and Human Society: The Quest for a Sustainable World; National Academy Press: Washington, DC, USA, 2000; pp. 325–336. [Google Scholar]
- Clavero, M.; García-Berthou, E. Invasive species are a leading cause of animal extinctions. Trends Ecol. Evol. 2005, 20, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everett, R.A. Patterns and pathways of biological invasions. Trends Ecol. Evol. 2000, 15, 177–178. [Google Scholar] [CrossRef]
- Pimentel, D.; McNair, S.; Janecka, J.; Wightman, J.; Simmonds, C.; O’connell, C.; Wong, E.; Russel, L.; Zern, J.; Aquino, T. Economic and environmental threats of alien plant, animal, and microbe invasions. Agric. Ecosyst. Environ. 2001, 84, 1–20. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, J.; Luo, Y.; Tao, J. The identification of risk sources of forestry invasive alien species in China and their controlling countermeasures. Plant Quar. 2015, 29, 42–46. [Google Scholar] [CrossRef]
- Lu, M.; Sun, J. Red Turpentine Beetle Dendroctonus valens LeConte. In Biological Invasions and Its Management in China; Wan, F., Jiang, M., Zhan, A., Eds.; Springer: Berlin, Germany, 2017; pp. 219–228. [Google Scholar]
- Cao, L.J.; Wei, S.J.; Hoffmann, A.A.; Wen, J.B.; Chen, M. Rapid genetic structuring of populations of the invasive fall webworm in relation to spatial expansion and control campaigns. Divers. Distrib. 2016, 22, 1276–1287. [Google Scholar] [CrossRef]
- Chen, Y.; Vasseur, L.; You, M. Potential distribution of the invasive loblolly pine mealybug, Oracella acuta (Hemiptera: Pseudococcidae), in Asia under future climate change scenarios. Clim. Chang. 2017, 141, 719–732. [Google Scholar] [CrossRef]
- Wei, S.-J.; Shi, B.-C.; Gong, Y.-J.; Jin, G.-H.; Chen, X.-X.; Meng, X.-F. Genetic structure and demographic history reveal migration of the diamondback moth Plutella xylostella (Lepidoptera: Plutellidae) from the southern to northern regions of China. PLoS ONE 2013, 8, e59654. [Google Scholar] [CrossRef]
- Wei, S.J.; Cao, L.J.; Gong, Y.J.; Shi, B.C.; Wang, S.; Zhang, F.; Guo, X.J.; Wang, Y.M.; Chen, X.X. Population genetic structure and approximate B ayesian computation analyses reveal the southern origin and northward dispersal of the oriental fruit moth G rapholita molesta (L epidoptera: T ortricidae) in its native range. Mol. Ecol. 2015, 24, 4094–4111. [Google Scholar] [CrossRef]
- Javal, M.; Roques, A.; Haran, J.; Hérard, F.; Keena, M.; Roux, G. Complex invasion history of the Asian long-horned beetle: Fifteen years after first detection in Europe. J. Pest Sci. 2019, 92, 173–187. [Google Scholar] [CrossRef]
- Downie, D. Locating the sources of an invasive pest, grape phylloxera, using a mitochondrial DNA gene genealogy. Mol. Ecol. 2002, 11, 2013–2026. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-M.; Sun, J.-T.; Xue, X.-F.; Li, J.-B.; Hong, X.-Y. Invasion genetics of the western flower thrips in China: Evidence for genetic bottleneck, hybridization and bridgehead effect. PLoS ONE 2012, 7, e34567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auger-Rozenberg, M.A.; Boivin, T.; Magnoux, E.; Courtin, C.; Roques, A.; Kerdelhué, C. Inferences on population history of a seed chalcid wasp: Invasion success despite a severe founder effect from an unexpected source population. Mol. Ecol. 2012, 21, 6086–6103. [Google Scholar] [CrossRef] [PubMed]
- Lippens, C.; Estoup, A.; Hima, M.; Loiseau, A.; Tatard, C.; Dalecky, A.; Bâ, K.; Kane, M.; Diallo, M.; Sow, A. Genetic structure and invasion history of the house mouse (Mus musculus domesticus) in Senegal, West Africa: A legacy of colonial and contemporary times. Heredity 2017, 119, 64–75. [Google Scholar] [CrossRef] [Green Version]
- Spradbery, J.; Kirk, A. Aspects of the ecology of siricid woodwasps (Hymenoptera: Siricidae) in Europe, North Africa and Turkey with special reference to the biological control of Sirex noctilio F. in Australia. Bull. Entomol. Res. 1978, 68, 341–359. [Google Scholar] [CrossRef] [Green Version]
- Boissin, E.; Hurley, B.; Wingfield, M.; Vasaitis, R.; Stenlid, J.; Davis, C.; De Groot, P.; Ahumada, R.; Carnegie, A.; Goldarazena, A. Retracing the routes of introduction of invasive species: The case of the Sirex noctilio woodwasp. Mol. Ecol. 2012, 21, 5728–5744. [Google Scholar] [CrossRef]
- Haugen, D.; Bedding, R.; Underdown, M.; Neumann, F. National Strategy for Control of Sirex noctilio in Australia; Australian Forest Growers: Canberra, Australia, 1990. [Google Scholar]
- Neumann, F.; Minko, G. The sirex wood wasp in Australian radiata pine plantations. Aust. For. 1981, 44, 46–63. [Google Scholar] [CrossRef]
- Li, D.; Shi, J.; Lu, M.; Ren, L.; Zhen, C.; Luo, Y. Detection and identification of the invasive Sirex noctilio (Hymenoptera: Siricidae) fungal symbiont, Amylostereum areolatum (Russulales: Amylostereacea), in China and the stimulating effect of insect venom on laccase production by A. areolatum YQL03. J. Econ. Entomol. 2015, 108, 1136–1147. [Google Scholar] [CrossRef]
- Sun, X.; Tao, J.; Ren, L.; Shi, J.; Luo, Y. Identification of Sirex noctilio (Hymenoptera: Siricidae) using a species-specific cytochrome C oxidase subunit I PCR assay. J. Econ. Entomol. 2016, 109, 1424–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Q. Pest Risk Analysis in China and the Economic Impacts Assessment of the Trade Measures for Sirex noctilio; Beijing Forestry University: Beijing, China, 2010. [Google Scholar]
- Schiff, N.M.; Goulet, H.; Smith, D.R.; Boudreault, C.; Wilson, A.D.; Scheffler, B.E. Siricidae (Hymenoptera: Symphyta: Siricoidea) of the western hemisphere. Can. J. Arthropod Identif. 2012, 21, 1–305. [Google Scholar]
- De Waard, J.R.; Ivanova, N.V.; Hajibabaei, M.; Hebert, P.; Martin, C. Methods in Molecular Biology: Environmental Genetics; Humana Press: Totowa, NJ, USA, 2008. [Google Scholar]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. Bioinform. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Leigh, J.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Bandelt, H.-J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Clement, M.J.; Snell, Q.; Walker, P.; Posada, D.; Crandall, K.A. TCS: Estimating gene genealogies. Parallel Distrib. Process. Symp. 2002, 2, 184. [Google Scholar]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. 2005, 1, 117693430500100003. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.; Zhu, J.; Li, Z.; Xu, M. Natural distribution of Pinus sylvestris var. mongolica on sandy land and its cultivation as an exotic species. Chin. J. Ecol. 2004, 23, 134–139. [Google Scholar] [CrossRef]
- Xu, Q.; Sun, X.-T.; Lu, P.-F.; Luo, Y.-Q.; Shi, J. Volatile profiles of three tree species in the northeastern China and associated effects on Sirex noctilio activity. J. Plant Interact. 2019, 14, 334–339. [Google Scholar] [CrossRef] [Green Version]
- Freeland, J.R.; Kirk, H.; Petersen, S. Molecular Ecology, 2nd ed.; Wiley: Hoboken, NJ, USA, 2011. [Google Scholar] [CrossRef]
- Lantschner, M.V.; Villacide, J.M.; Garnas, J.R.; Croft, P.; Carnegie, A.J.; Liebhold, A.M.; Corley, J.C. Temperature explains variable spread rates of the invasive woodwasp Sirex noctilio in the Southern Hemisphere. Biol. Invasions 2014, 16, 329–339. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zheng, Z.; Zhou, F.; Ren, L.; Luo, Y. Effects of age (in days), gender and nematode infection on the flight capacity of Sirex noctilio. Chin. J. Appl. Entomol. 2017, 54. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locality | Population Code | First Detection | Sampling Time | N(Sequenced Individuals) | N variable Sites | Nht | h | π |
|---|---|---|---|---|---|---|---|---|
| Total | 364(778) | |||||||
| Asia, China | 193(607) | 7 | 8 | 0.311 | 0.00114 | |||
| Dumeng, Heilongjiang | DM | 2013 | 2016 | 20(66) | 2 | 3 | 0.549 | 0.00183 |
| Qiqihaer, Heilongjiang | 2013 | 2016 | 20(64) | 1 | 2 | 0.185 | 0.00057 | |
| Hegang, Heilongjiang | HG | 2014 | 2015-16 | 20(204) | 2 | 3 | 0.472 | 0.00191 |
| Fujin, Heilongjiang | FJ | 2014 | 2015 | 20(69) | 2 | 3 | 0.344 | 0.00111 |
| Hulunbeier, Inner Mongolia | HHEJ, HLBE | 2014 | 2014 | 8 | 1 | 2 | 0.4 | 0.00124 |
| Keyihe, Inner Mongolia | HDQ | 2013 | 2013 | 1 | 0 | 1 | 0 | 0 |
| Yushu, Jilin | YS | 2015 | 2015 | 14 | 1 | 2 | 0.138 | 0.00043 |
| Jinbaotun, Inner Mongolia | JBT | 2015 | 2015-16 | 20(83) | 0 | 1 | 0 | 0 |
| Changtu, Liaoning | CT | 2016 | 2016 | 20(48) | 0 | 1 | 0 | 0 |
| Changchun, Jilin | CC | 2013 | 2013 | 1 | 0 | 1 | 0 | 0 |
| Zhanggutai, Liaoning | ZGT | 2016 | 2016 | 18 | 0 | 1 | 0 | 0 |
| Kangping, Liaoning | KP | 2016 | 2016 | 18 | 0 | 1 | 0 | 0 |
| Chifeng, Inner Mongolia | CF | 2016 | 2016 | 2 | 0 | 1 | 0 | 0 |
| Acheng, Heilongjiang | AC | 2013 | 2013 | 1 | 0 | 1 | 0 | 0 |
| Mudanjiang, Heilongjiang | MDJ | 2013 | 2013 | 6 | 0 | 1 | 0 | 0 |
| Ningan, Heilongjiang MDJ-DJ | 2013 | |||||||
| Suiyang, Heilongjiang MDJ-SY | 2013 | |||||||
| Muleng, Heilongjiang MDJ-BMT | 2013 | |||||||
| Hailin, Heilongjiang MDJ-CH | 2013 | |||||||
| Mudanjiang, Heilongjiang MDJ-SGLK | 2013 | |||||||
| Mudanjiang, Heilongjiang MDJ-XS | 2013 | |||||||
| Harbin, Heilongjiang | XL | 2013 | 2013 | 1 | 0 | 1 | 0 | 0 |
| Nongan, Jilin | NAN | 2014 | 2014 | 1 | 0 | 1 | 0 | 0 |
| Jiamusi, Heilongjiang | HNC | 2013 | 2013 | 1 | 0 | 1 | 0 | 0 |
| Manzhouli, Inner Mongolia | MZL | 2013 | 2013 | 1 | 0 | 1 | 0 | 0 |
| Africa | 22 | 0 | 1 | 0 | 0 | |||
| South Africa | SA | 1 | 0 | 1 | 0 | 0 | ||
| Natal Midlands | NM | 13 | 0 | 1 | 0 | 0 | ||
| Western Cape | WC | 6 | 0 | 1 | 0 | 0 | ||
| Eastern Cape | EC | 2 | 0 | 1 | 0 | 0 | ||
| North America | 39 | 1 | 2 | 0.264 | 0.00082 | |||
| Canada | Can | 27 | 1 | 2 | 0.352 | 0.00109 | ||
| Canada-DRYAD | Can | |||||||
| USA | NY, USA | 12 | 0 | 1 | 0 | 0 | ||
| USA-DRYAD | USA | 0 | 1 | 0 | 0 | |||
| South America | 50 | 2 | 3 | 0.563 | 0.00202 | |||
| Argentina (unknow) | Arg | 25 | 2 | 3 | 0.568 | 0.00237 | ||
| Arg-DRYAD | Arg, ArgES | |||||||
| Arg-Arroyo del medio | Arg | 2018 | ||||||
| Arg-Aluminé | Arg | 2018 | ||||||
| Arg-Andacollo | Arg | 2019 | ||||||
| Uruguay | Uruguay | 1 | 0 | 1 | 0 | 0 | ||
| Chile | Chi, Chil | 24 | 1 | 2 | 0.511 | 0.00159 | ||
| Chil-DRYAD | Chi, ChiS | |||||||
| Oceania | 16 | 1 | 2 | 0.226 | 0.0007 | |||
| Australia | Austra, Austr | 16 | 1 | 2 | 0.226 | 0.0007 | ||
| Austr-DRYAD | AustrES, AustrEB | |||||||
| Europe | 44 | 4 | 4 | 0.533 | 0.00194 | |||
| Switzerland | Switz | 15 | 0 | 1 | 0 | 0 | ||
| Swit-DRYAD | Swit, SW, Swi | |||||||
| Italy | Ita-XS | 1 | 0 | 1 | 0 | 0 | ||
| Poland | Pol-XS | 1 | 0 | 1 | 0 | 0 | ||
| Hungary | Hun, Hun-XS | 2018 | 4,2 | 0 | 1 | 0 | 0 | |
| Ger-Berlin | Ger-Ber | 2 | 0 | 1 | 0 | 0 | ||
| Ger-Stuttgart | Ger-Stu | 1 | 0 | 1 | 0 | 0 | ||
| Spain | Spain | 3 | 0 | 1 | 0 | 0 | ||
| Spain-DRYAD | Spain | |||||||
| Fr-Labenne | Fr6-Lab., Fr-Lab.XS30 | 2017 | 1,1 | 0 | 1 | 0 | 0 | |
| Fr-Meolans Revel | Fr03-MR | 1 | 0 | 1 | 0 | 0 | ||
| Fr-Villes sur Auzon | Fr01-V.S.A | 1 | 0 | 1 | 0 | 0 | ||
| Fr-Marseille | Fr3-Mar. | 2013 | 1 | 0 | 1 | 0 | 0 | |
| Fr-Tarascon | Fr11-Tara | 2017 | 1 | 0 | 1 | 0 | 0 | |
| Fr-Brain sur Allones | Fr8-B.S.A., Fr-B.S.A.XS31 | 2017 | 1,1 | 1 | 2 | 0.667 | 0.00207 | |
| Fr-Etaules | Fr4-Eta. | 2017 | 1 | 0 | 1 | 0 | 0 | |
| Fr-Lavercantière | Fr18-Lav. | 2011 | 1 | 0 | 1 | 0 | 0 | |
| Fr-Orléans | Fr21-Orle, Fr87-Orle | 2017.2 | 2 | 1 | 2 | 0.667 | 0.00207 | |
| Fr-St Alban des Hurtiè | Fr16-St.Al | 2017 | 1 | 0 | 1 | 0 | 0 | |
| Fr-Roissy CDG | Fr33-CDG | 2017 | 1 | 0 | 1 | 0 | 0 | |
| Fr-Tartigny | Fr12-Tart | 2016 | 1 | 0 | 1 | 0 | 0 |
| Source of Variation | d.f. | Sum of Squares | Variance Components | Percentage of Variation | P |
|---|---|---|---|---|---|
| AMOVA—Chinese populations grouped by geographic proximity (West-East) | |||||
| Among groups | 3 | 11.561 | 0.08286 | 38.64 *** | 0.006 |
| Among populations within groups | 15 | 4.256 | 0.01961 | 9.15 *** | 0.003 |
| Within populations | 174 | 19.479 | 0.11195 | 52.21 *** | 0.000 |
| AMOVA—Chinese populations grouped by latitude (North-South) | |||||
| Among groups | 1 | 6.262 | 0.05510 | 25.58 *** | 0.004 |
| Among populations within groups | 17 | 9.555 | 0.04838 | 22.46 *** | 0.000 |
| Within populations | 174 | 19.479 | 0.11195 | 51.97 *** | 0.000 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, X.; Tao, J.; Roques, A.; Luo, Y. Invasion History of Sirex noctilio Based on COI Sequence: The First Six Years in China. Insects 2020, 11, 111. https://doi.org/10.3390/insects11020111
Sun X, Tao J, Roques A, Luo Y. Invasion History of Sirex noctilio Based on COI Sequence: The First Six Years in China. Insects. 2020; 11(2):111. https://doi.org/10.3390/insects11020111
Chicago/Turabian StyleSun, Xueting, Jing Tao, Alain Roques, and Youqing Luo. 2020. "Invasion History of Sirex noctilio Based on COI Sequence: The First Six Years in China" Insects 11, no. 2: 111. https://doi.org/10.3390/insects11020111