Descriptive Bacterial and Fungal Characterization of Propolis Using Ultra-High-Throughput Marker Gene Sequencing




Abstract
:1. Introduction
2. Materials and Methods
2.1. Propolis Samples
2.2. Total Genomic DNA Extraction
2.3. Analysis of Bacterial Microbiota
2.4. Analysis of Fungal Microbiota
2.5. Microbial Diversity Analyses
3. Results
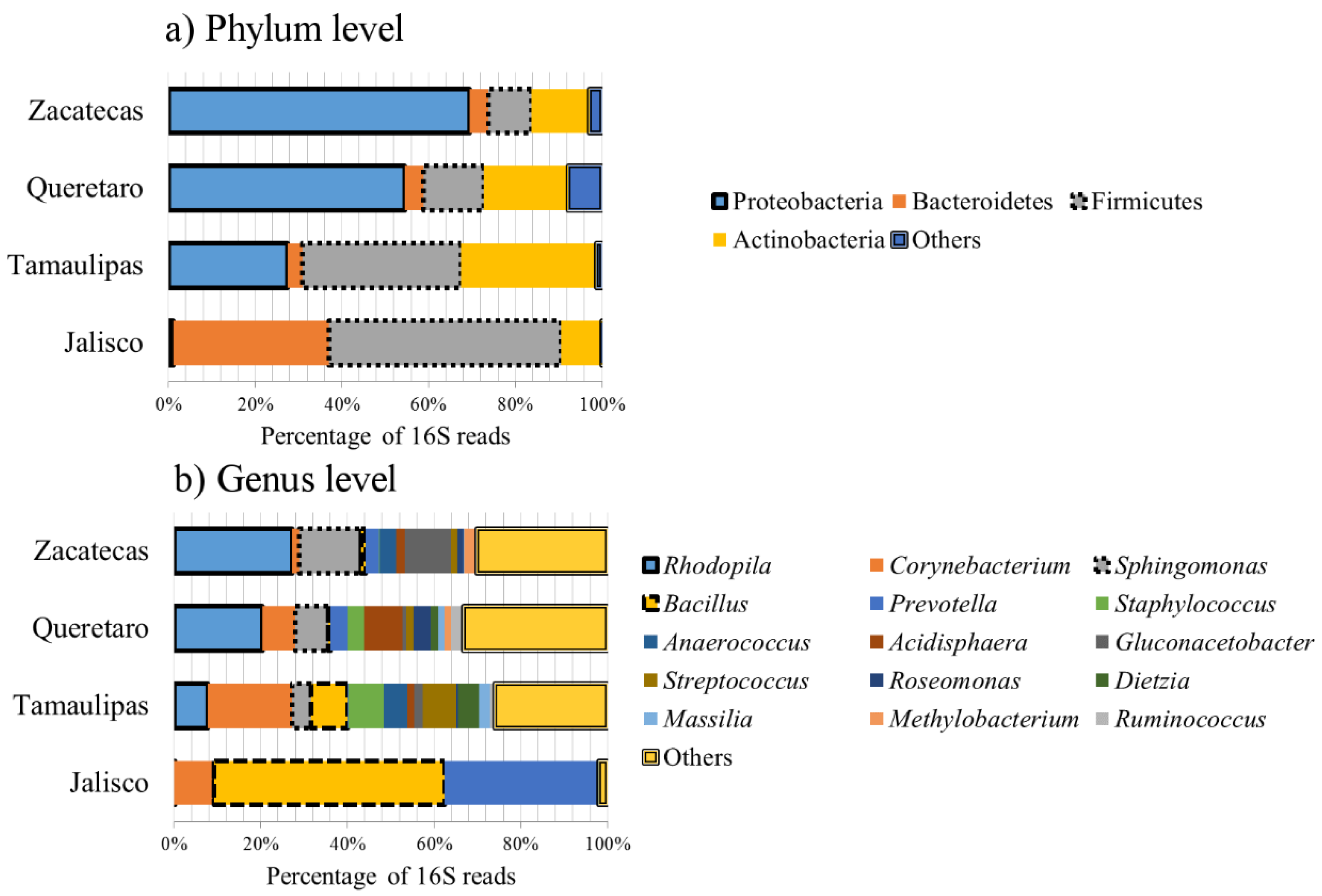
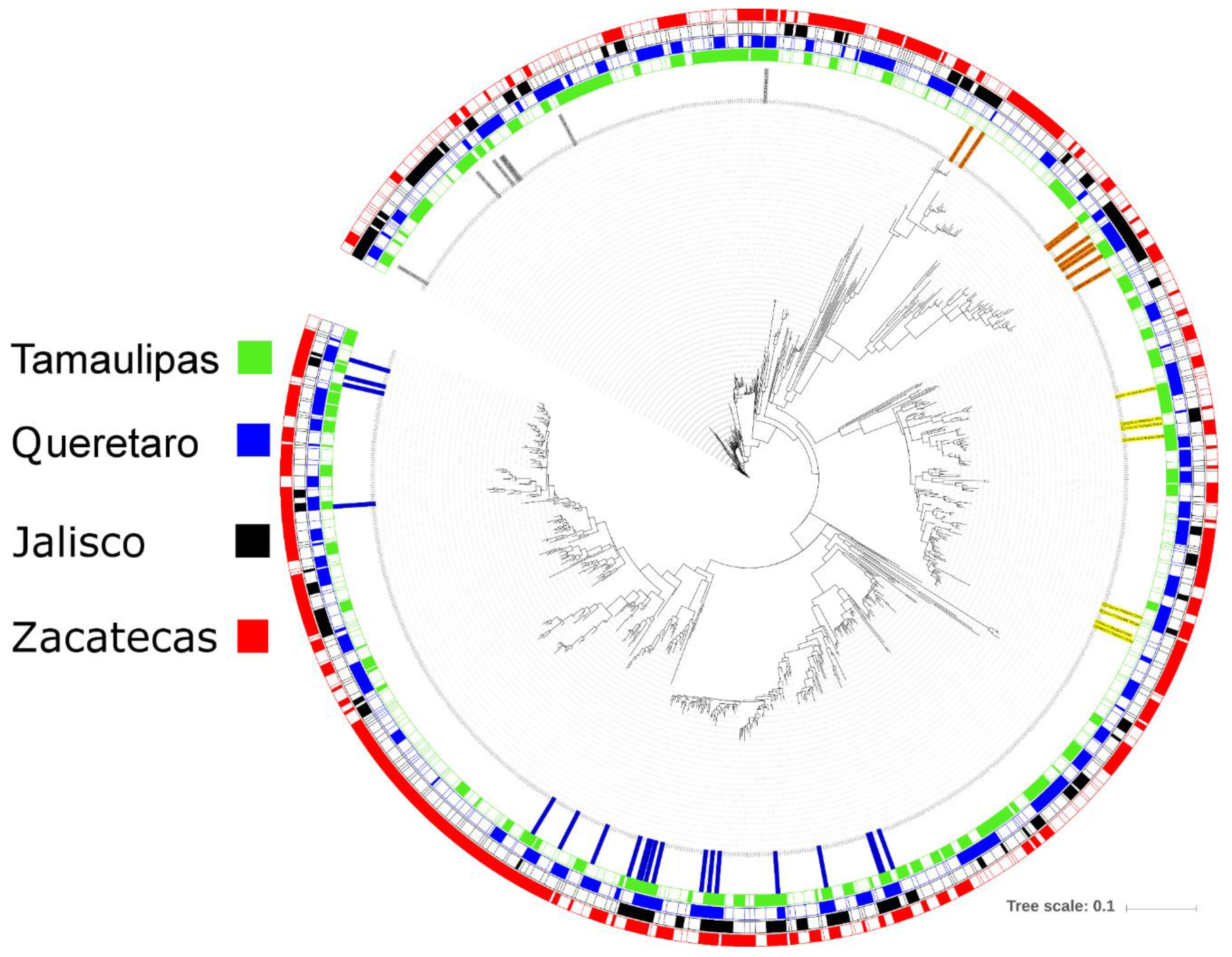
3.1. Bacterial Microbiota
3.2. Predicted Metabolic Profile of Bacterial Microbiota
3.3. Fungal Microbiota
3.4. Microbial Diversity
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kumazawa, S.; Yoneda, M.; Shibata, I.; Kanaeda, J.; Hamasaka, T.; Nakayama, T. Direct evidence for the plant origin of Brazilian propolis by the observation of honeybee behavior and phytochemical analysis. Chem. Pharm. Bull. 2003, 51, 740–742. [Google Scholar] [CrossRef] [PubMed]
- Borba, R.S.; Klyczek, K.K.; Mogen, K.L.; Spivak, M. Seasonal benefits of a natural propolis envelope to honeybee immunity and colony health. J. Experim. Biol. 2015, 218, 3689–3699. [Google Scholar] [CrossRef] [PubMed]
- Toreti, V.C.; Sato, H.H.; Pastore, G.M.; Park, Y.K. Recent progress of propolis for its biological and chemical compositions and its botanical origin. Evid. Based Complement. Altern. Med. 2013, 2013, 697390. [Google Scholar] [CrossRef] [PubMed]
- Silva-Carvalho, R.; Baltazar, F.; Almeida-Aguiar, C. Propolis: A complex natural product with a plethora of biological activities that can be explored for drug development. Evid. Based Complement. Altern. Med. 2015, 2015, 206439. [Google Scholar] [CrossRef] [PubMed]
- Sforcin, J.M. Propolis and the immune system: A review. J. Ehtnopharmacol. 2007, 113, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kujumgiev, A.; Tsvetkova, I.; Serkedjieva, Y.; Bankova, V.; Christov, R.; Popov, S. Antibacterial, antifungal and antiviral activity of propolis of different geographic origin. J. Ethnopharmacol. 1999, 64, 235–240. [Google Scholar] [CrossRef]
- Martinson, V.G.; Danforth, B.N.; Minckley, R.L.; Rueppell, O.; Tingek, S.; Moran, N.A. A simple and distinctive microbiota associated with honeybees and bumble bees. Mol. Ecol. 2011, 20, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Engel, P.; Kwong, W.K.; McFrederick, Q.; Anderson, K.E.; Barribeau, S.M.; Chandler, J.A.; Cornman, R.S.; Dainat, J.; de Miranda, J.R.; Doublet, V.; et al. The bee microbiome: Impact on bee health and model for evolution and ecology of host-microbe interactions. mBio 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.J.F.; Paxton, R.J. The conservation of bees: A global perspective. Apidologie 2009, 40, 410–416. [Google Scholar] [CrossRef]
- Douglas, A.E. Multiorganismal insects: Diversity and function of resident microorganisms. Ann. Rev. Entomol. 2015, 60, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.E.; Sheehan, T.H.; Mott, B.M.; Maes, P.; Snyder, L.; Schwan, M.R.; Walton, A.; Jones, B.M.; Corby-Harris, V. Microbial ecology of the hive and pollination landscape: Bacterial associated from floral nectar, the alimentary tract and stored food of honeybees (Apis mellifera). PLoS ONE 2013, 8, e83125. [Google Scholar] [CrossRef] [PubMed]
- Grubbs, K.J.; Scott, J.J.; Budsberg, K.J.; Read, H.; Balser, T.C.; Currie, C.R. Unique honeybee (Apis mellifera) hive component-based communities as detected by a hybrid of phospholipid fatty-acid and fatty-acid methyl ester analyses. PLoS ONE 2015, 10, e0121697. [Google Scholar] [CrossRef]
- Willers, C.; Jansen van Rensburg, P.J.; Claassens, S. Microbial signature lipid biomarker analysis—An approach that is still preferred, even amid various method modifications. J. Appl. Microbiol. 2015, 118, 1251–1263. [Google Scholar] [CrossRef] [PubMed]
- Ngalimat, M.S.; Raja, A.; Rahman, R.N.Z.; Yusof, M.T.; Syahir, A.; Sabri, S. Characterisation of bacteria isolated from the stingless bee, Heterotrigona itama, honey, bee bread and propolis. Peer J. 2019, 7, e7478. [Google Scholar] [CrossRef] [PubMed]
- Reybroeck, W.; Daeseleire, E.; De Brabander, H.F.; Herman, L. Antimicrobials in beekeeping. Vet. Microbiol. 2012, 158, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.A.; Marchioro, G.M.; Costa, A.C.; Araujo, E.D. Extraction and amplification of DNA from Brazilian red propolis. Scientia Plena 2014, 10, 039902. [Google Scholar]
- Soares, S.; Amaral, J.S.; Oliveira, M.B.P.P.; Mafra, I. Improving DNA isolation from honey for the botanical origin identification. Food Control 2015, 48, 130–136. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Mazcorro, J.F.; Pedreschi, R.; Yuan, J.; Kawas, J.R.; Chew, B.; Dowd, S.E.; Noratto, G. Apple consumption is associated with a distinctive microbiota, proteomics and metabolomics profile in the gut of Dawley Sprague rats fed a high-fat diet. PLoS ONE 2019, 14, e0212586. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Mazcorro, J.F.; Pedreschi, R.; Chew, B.; Dowd, S.E.; Kawas, J.R.; Noratto, G. Dietary supplementation with raspberry extracts modifies the fecal microbiota in obese diabetic db/db mice. J. Microbiol. Biotechnol. 2018, 28, 1247–1259. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Gonzalez Peña, A.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Meth. 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Snisky, J.J., White, T.J., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 315–322. [Google Scholar]
- Lutzoni, F.; Kauff, F.; Cox, C.J.; McLaughlin, D.; Celio, G.; Dentinger, B.; Padamsee, M.; Hibbett, D.; James, T.Y.; Baloch, E.; et al. Assembling the fungal tree of life: Progress, classification, and evolution of subcellular traits. Am. J. Bot. 2004, 91, 1446–1480. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Fungal Barcoding Consortium. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.H.; Hyde, K.; Pawlowska, J.; Ryberg, M.; Tedersoo, L.; Bjørnsgard, A.; Alias, S.A.; Alves, A.; Anderson, C.L.; Antonelli, A.; et al. Improving ITS sequence data for identification of plant pathogenic fungi. Fungal Divers. 2014, 67, 11–19. [Google Scholar] [CrossRef]
- Findley, K.; Oh, J.; Yang, J.; Conlan, S.; Deming, C.; Meyer, J.A.; Schoenfeld, D.; Nomicos, E.; Park, M.; NISC Comparative Sequencing Program; et al. Human skin fungal diversity. Nature 2013, 498, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Bent, S.J.; Forney, L.J. The tragedy of the uncommon: Understanding limitations in the analysis of microbial diversity. ISME J. 2008, 2, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Benning, M.M.; Meyer, T.E.; Holden, H.M. Molecular structure of a high potential cytochrome c2 isolated from Rhodopila globiformis. Arch. Biochem. Biophys. 1996, 333, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Kwong, W.K.; Moran, N.A. Evolution of host specialization in gut microbes: The bee gut as a model. Gut Microbes 2015, 6, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Kwong, W.K.; Moran, N.A. Cultivation and characterization of the gut symbionts of honeybees and bumble bees: Description of Snodgrassella alvi gen. nov., sp. nov., a member of the family Neisseriaceae of the Betaproteobacteria, and Gilliamella apicola gen. nov., sp. nov., a member of Orbaceae fam. nov., Orbales ord. nov., a sister taxon to the order ‘Enterobacteriales’ of the Gammaproteobacteria. Int. J. Syst. Evol. Microbiol. 2013, 63, 2008. [Google Scholar] [CrossRef] [PubMed]
- Kagle, J.; Porter, A.W.; Murdoch, R.W.; Rivera-Cancel, G.; Hay, A.G. Biodegradation of pharmaceutical and personal care products. Adv. Appl. Microbiol. 2009, 67, 65–108. [Google Scholar] [CrossRef] [PubMed]
- Kolvenbach, B.A.; Corvini, P.F. The degradation of alkylphenols by Sphingomonas sp. strain TTNP3—A review on seven years of research. New Biotechnol. 2012, 30, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Parshikov, I.A.; Silva, E.O.; Furtado, N.A. Transformation of saturated nitrogen-containing heterocyclic compounds by microorganisms. Appl. Microbiol. Biotechnol. 2014, 98, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Bittinger, K.; Bushman, F.D.; DeSantis, T.Z.; Andersen, G.L.; Knight, R. PyNAST: A flexible tool for aligning sequences to a template alignment. Bioinformatics 2010, 26, 266–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v4: Recent updates and new developments. Nucl. Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, X. DAMBE5: A comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2013, 30, 1720–1728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackwell, M. The fungi: 1, 2, 3…5.1 million species? Am. J. Bot. 2011, 98, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Wahdan, H.A. Causes of the antimicrobial activity of honey. Infection 1998, 26, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, T.C.; Butler, E.; Markowicz, P.; Lindholm, C.; Larsson, L.; Vásquez, A. Lactic acid bacterial symbionts in honeybees—An unknown key to honey’s antimicrobial and therapeutic activities. Int. Wound J. 2016, 13, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Ben-Amor, K.; Heilig, H.; Smidt, H.; Vaughan, E.E.; Abee, T.; de Vos, W.M. Genetic diversity of viable, injured, and dead fecal bacteria assessed by fluorescence-activated cell sorting and 16S rRNA gene analysis. Appl. Environ. Microbiol. 2005, 71, 4679–4689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, J.E.; Martinson, V.G.; Urban-Mead, K.; Moran, N.A. Routes of acquisition of the gut microbiota of the honeybee Apis mellifera. Appl. Environ. Microbiol. 2014, 80, 7378–7387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berdoulay, M.; Salvado, J.C. Genetic characterization of microbial communities living at the surface of building stones. Lett. Appl. Microbiol. 2009, 49, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Bragina, A.; Maier, S.; Berg, C.; Müller, H.; Chobot, V.; Hadacek, F.; Berg, G. Similar diversity of Alphaproteobacteria and nitrogenase gene amplicons on two related sphagnum mosses. Front. Microbiol. 2012, 2, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogales, B.; Moore, E.R.; Abraham, W.R.; Timmis, K.N. Identification of the metabolically active members of a bacterial community in a polychlorinated biphenyl-polluted moorland soil. Environ. Microbiol. 1999, 1, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Yabuuchi, E.; Yano, I.; Oyaizu, H.; Hashimoto, Y.; Ezaki, T.; Yamamoto, H. Proposals of Sphingomonas paucimobilis gen. nov. and comb. nov., Sphingomonas parapaucimobilis sp. nov., Sphingomonas yanoikuyae sp. nov., Sphingomomnas adhaesiva sp. nov., Sphingomonas capsulate comb. nov., and two genospecies of the genus Sphingomonas. Microbiol. Immunol. 1990, 34, 99–119. [Google Scholar] [CrossRef] [PubMed]
- Dezmirean, D.S.; Mărghitaş, L.A.; Chirilă, F.; Copaciu, F.; Simonca, V.; Bobiş, O.; Erler, S. Influence of geographic origin, plant source and polyphenolic substances on antimicrobial properties of propolis against human and honeybee pathogens. J. Apic. Res. 2017, 56, 588–597. [Google Scholar] [CrossRef]
- Halwachs, B.; Madhusudhan, N.; Krause, R.; Henrik Nilsson, R.; Moissl-Eichinger, C.; Högenauer, C.; Thallinger, G.G.; Gorkiewicz, G. Critical issues in mycobiota analysis. Front. Microbiol. 2017, 8, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vega, F.E.; Dowd, P.F. The role of yeast as insect endosymbionts. In Insect-Fungal Associations: Ecology and Evolution; Vega, F.E., Blackwell, M., Eds.; Oxford University Press: New York, NY, USA, 2005; pp. 211–243. [Google Scholar]
- Irish, J.; Carter, D.A.; Shokohi, T.; Blair, S.E. Honey has an antifungal effect against Candida species. Med. Mycol. 2006, 44, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Koç, A.N.; Silici, S.; Kasap, F.; Hörmet-Oz, H.T.; Mavus-Buldu, H.; Ercal, B.D. Antifungal activity of the honeybee products against Candida spp. and Trichosporon spp. J. Med. Food 2011, 14, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Englezos, V.; Cocolin, L.; Rantsiou, K.; Ortiz-Julien, A.; Bloem, A.; Dequin, S.; Camarasa, C. Specific phenotypic traits of Starmerella bacillaris regarding nitrogen source consumption and central carbon metabolites production during wine fermentation. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixeira, A.C.; Marini, M.M.; Nicoli, J.R.; Antonini, Y.; Martins, R.P.; Lachance, M.A.; Rosa, C.A. Starmerella meliponinorum sp. nov., a novel ascomycetous yeast species associated with stingless bees. Int. J. Syst. Evolut. Microbiol. 2003, 53, 330–343. [Google Scholar] [CrossRef] [PubMed]
- Rosa, C.A.; Lachance, M.A.; Silva, J.O.; Teixeira, A.C.; Marini, M.M.; Antonini, Y.; Martins, R.P. Yeast communities associated with stingless bees. FEMS Yeast Res. 2003, 4, 271–275. [Google Scholar] [CrossRef]
- Meason-Smith, C.; Diesel, A.; Patterson, A.P.; Older, C.E.; Mansell, J.M.; Suchodolski, J.S.; Rodrigues Hoffmann, A. What is living on your dog’s skin? Characterization of the canine cutaneous mycobiota and fungal dysbiosis in canine allergic dermatitis. FEMS Microbiol. Ecol. 2015, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| State | Location | Principal Flora |
|---|---|---|
| Jalisco | Santa Anita, Tlaquepaque LAT/LONG 20.55/103.45 | Blue maguey (Agave tequilana) and mesquite (Prosopis granulosa) |
| Queretaro | Conca, Arroyo Seco LAT/LONG 21.43/99.67 | Coffee (Coffea arabica) and mesquite (Prosopis granulosa) |
| Tamaulipas | El Carmen, Guemez LAT/LONG 24.07/99.13 | Orange trees (Citrus sinensis) and mesquite (Prosopis granulosa) |
| Zacatecas | Fresnillo LAT/LONG 32.17/102.88 | Mesquite (Prosopis granulosa) |
| Bacterial Group | Jalisco | Queretaro | Tamaulipas | Zacatecas | Overall Median |
|---|---|---|---|---|---|
| Rhodopila | 0.08% | 20.3% | 7.8% | 27.3% | 14.1% |
| Corynebacterium | 9.1% | 7.7% | 19.5% | 1.6% | 8.4% |
| Sphingomonas | 0.03% | 7.7% | 4.3% | 14.3% | 5.9% |
| Bacillus | 53.3% | 0.5% | 8.5% | 1.0% | 4.8% |
| Prevotella | 35.3% | 4.1% | 0.1% | 3.1% | 3.5% |
| Staphylococcus | 0.06% | 3.9% | 8.2% | 0.1% | 1.9% |
| Anaerococcus | 0.03% | 0.01% | 5.5% | 3.9% | 1.9% |
| Acidisphaera | 0.01% | 8.8% | 1.5% | 1.9% | 1.6% |
| Streptococcus | 0.02% | 1.6% | 7.8% | 1.5% | 1.5% |
| Gluconacetobacter | 0.02% | 1.0% | 2.1% | 10.8% | 1.5% |
| Roseomonas | 0.01% | 4.1% | 0.3% | 1.5% | 0.9% |
| Dietzia | 0.01% | 1.8% | 4.8% | 0.00% | 0.9% |
| Massilia | - | 1.4% | 2.6% | 0.2% | 0.8% |
| Methylobacterium | 0.00% | 2.9% | 1.1% | 0.4% | 0.8% |
| Ruminococcus | 0.00% | 2.9% | 1.1% | 0.4% | 0.8% |
| Ornithinibacillus | 0.01% | 1.3% | 1.7% | 0.00% | 0.67% |
| Microbacterium | 0.00% | 1.3% | - | 1.3% | 0.62% |
| Curvibacter | 0.50% | 0.80% | 0.53% | 0.57% | 0.55% |
| Clostridium | 0.01% | 0.82% | 1.7% | 0.05% | 0.44% |
| Flavobacterium | 0.50% | 0.31% | 1.9% | 0.31% | 0.41% |
| Curtobacterium | 0.03% | 5.3% | 0.01% | 0.71% | 0.37% |
| Pseudomonas | 0.02% | 3.5% | 0.58% | 0.08% | 0.33% |
| Cronobacter | 0.01% | 0.60% | 3.2% | 0.05% | 0.32% |
| Aquabacterium | 0.01% | 0.30% | 0.34% | 0.43% | 0.32% |
| Terriglobus | 0.02% | 5.5% | 0.01% | 0.47% | 0.24% |
| All other taxa | ~1% | ~13% | ~16% | ~25% | NA |
| Feature in PICRUSt | Jalisco | Queretaro | Tamaulipas | Zacatecas | Overall Median |
|---|---|---|---|---|---|
| Membrane transporters | 5.5% | 7.0% | 7.3% | 7.1% | 7.0% |
| ABC transporters | 3.0% | 4.2% | 4.3% | 4.3% | 4.3% |
| Ribosome | 2.7% | 2.1% | 2.3% | 2.0% | 2.2% |
| Peptidases | 2.3% | 1.8% | 1.9% | 1.7% | 1.8% |
| Pyrimidine metabolism | 2.1% | 1.6% | 1.8% | 1.5% | 1.7% |
| Transcription machinery | 1.3% | 0.9% | 0.9% | 0.8% | 0.9% |
| Starch and sucrose metabolism | 1.1% | 0.8% | 0.8% | 0.7% | 0.8% |
| Butanoate metabolism | 0.9% | 1.2% | 1.2% | 1.4% | 1.2% |
| DNA repair and recombination proteins | 3.3% | 2.9% | 3.0% | 2.8% | 3.0% |
| Secretion system | 1.4% | 1.7% | 1.8% | 1.7% | 1.7% |
| Chromosome | 1.7% | 1.4% | 1.5% | 1.4% | 1.5% |
| DNA replication proteins | 1.3% | 1.0% | 1.1% | 1.0% | 1.1% |
| Amino-acid-related enzymes | 1.7% | 1.4% | 1.5% | 1.4% | 1.5% |
| Phosphotransferase system | 0.2% | 0.3% | 0.5% | 0.2% | 0.2% |
| Cysteine and methionine metabolism | 1.2% | 1.0% | 1.0% | 1.0% | 1.0% |
| Peptidoglycan biosynthesis | 1.0% | 0.7% | 0.8% | 0.7% | 0.7% |
| Glutathione metabolism | 0.3% | 0.6% | 0.5% | 0.6% | 0.5% |
| Bacterial secretion system | 0.6% | 0.9% | 0.9% | 0.9% | 0.9% |
| Valine, leucine and isoleucine degradation | 0.8% | 1.0% | 0.9% | 1.2% | 1.0% |
| Two-component system (signal transduction) | 1.9% | 2.2% | 2.1% | 2.1% | 2.1% |
| Fungal Group | Jalisco | Queretaro | Tamaulipas | Zacatecas | Overall Median |
|---|---|---|---|---|---|
| Candida | 42.6% | 9.5% | 22.8% | 2.1% | 16.2% |
| Acremonium | 0.03% | 25.2% | 10.6% | 2.6% | 6.6% |
| Aspergillus | 0.08% | 6.6% | 42.6% | 2.2% | 4.4% |
| Phanerochaete | 0.11% | 22.3% | 3.6% | 2.9% | 3.2% |
| Cladosporium | 1.3% | 4.9% | 0.65% | 2.8% | 2.1% |
| Starmerella | 42.5% | 1.9% | 2.1% | 0.3% | 2.0% |
| Monascus | 0.11% | 7.2% | 2.8% | 0.41% | 1.6% |
| Wallemia | 0.11% | 1.1% | 1.7% | 1.2% | 1.2% |
| Alternaria | - | 0.39% | 2.8% | 1.0% | 0.71% |
| Malassezia | - | 0.01% | 2.1% | 1.13% | 0.57% |
| Trichosporonoides | - | 4.2% | 0.30% | 0.53% | 0.42% |
| Moniliella | 12.9% | 0.76% | 0.06% | 0.04% | 0.41% |
| Phoma | - | 0.03% | 0.78% | 17.6% | 0.40% |
| Penicillium | - | 0.37% | 3.2% | 0.06% | 0.21% |
| Cephalosporium | 0.06% | 11.6% | 0.05% | 0.34% | 0.20% |
| Ampelomyces | - | - | 0.34% | 0.47% | 0.17% |
| Coniochaeta | 0.08% | 0.03% | 0.04% | 43.8% | 0.06% |
| Spencermartinsia | 0.06% | 0.01% | 3.2% | 0.07% | 0.06% |
| Ramulispora | - | 1.8% | 0.01% | 0.11% | 0.06% |
| Priceomyces | - | 0.18% | - | 0.05% | 0.02% |
| Hyphopichia | 0.03% | - | 0.17% | 0.02% | 0.02% |
| Debaryomyces | 0.03% | - | 0.12% | 0.02% | 0.02% |
| Batcheloromyces | 0.00% | 0.95% | 0.00% | 0.04% | 0.02% |
| Aureobasidium | 0.03% | 0.01% | 0.01% | 10.2% | 0.02% |
| Eremascus | - | 0.85% | 0.01% | 0.02% | 0.01% |
| Exophiala | 0.03% | - | - | 0.92% | 0.01% |
| Dactylella | 0.03% | - | - | 2.5% | 0.01% |
| Knufia | 0.03% | - | - | 1.6% | 0.01% |
| Sclerostagonospora | - | - | 0.01% | 0.80% | 0.01% |
| Cyphellophora | - | 0.00% | - | 0.68% | 0.00% |
| Westerdykella | - | - | - | 0.45% | 0.00% |
| Kwoniella | - | - | - | 1.06% | 0.00% |
| Fusarium | - | 0.14% | - | - | 0.00% |
| Curvularia | - | - | - | 0.10% | 0.00% |
| Mucor | - | - | - | 0.37% | 0.00% |
| Cryptococcus | - | - | - | 0.06% | 0.00% |
| Zeloasperisporium | - | - | - | 1.6% | 0.00% |
| Sample | OTUs | Chao1 | Shannon | Simpson |
|---|---|---|---|---|
| Jalisco (fungal) | 13 | 20 | 1.1 | 0.51 |
| Tamaulipas (fungal) | 28 | 56 | 2.2 | 0.65 |
| Queretaro (fungal) | 33 | 35 | 2.8 | 0.81 |
| Zacatecas (fungal) | 37 | 39 | 2.6 | 0.69 |
| Jalisco (bacterial) | 240 | 510 | 1.9 | 0.61 |
| Tamaulipas (bacterial) | 357 | 663 | 5.6 | 0.95 |
| Queretaro (bacterial) | 363 | 611 | 5.1 | 0.90 |
| Zacatecas (bacterial) | 542 | 1048 | 6.0 | 0.92 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia-Mazcorro, J.F.; Kawas, J.R.; Marroquin-Cardona, A.G. Descriptive Bacterial and Fungal Characterization of Propolis Using Ultra-High-Throughput Marker Gene Sequencing. Insects 2019, 10, 402. https://doi.org/10.3390/insects10110402
Garcia-Mazcorro JF, Kawas JR, Marroquin-Cardona AG. Descriptive Bacterial and Fungal Characterization of Propolis Using Ultra-High-Throughput Marker Gene Sequencing. Insects. 2019; 10(11):402. https://doi.org/10.3390/insects10110402
Chicago/Turabian StyleGarcia-Mazcorro, Jose F., Jorge R. Kawas, and Alicia G. Marroquin-Cardona. 2019. "Descriptive Bacterial and Fungal Characterization of Propolis Using Ultra-High-Throughput Marker Gene Sequencing" Insects 10, no. 11: 402. https://doi.org/10.3390/insects10110402