Case of Early-Onset Parkinson’s Disease in a Heterozygous Mutation Carrier of the ATP7B Gene

 and
and
Abstract
:1. Introduction
2. Materials and Methods
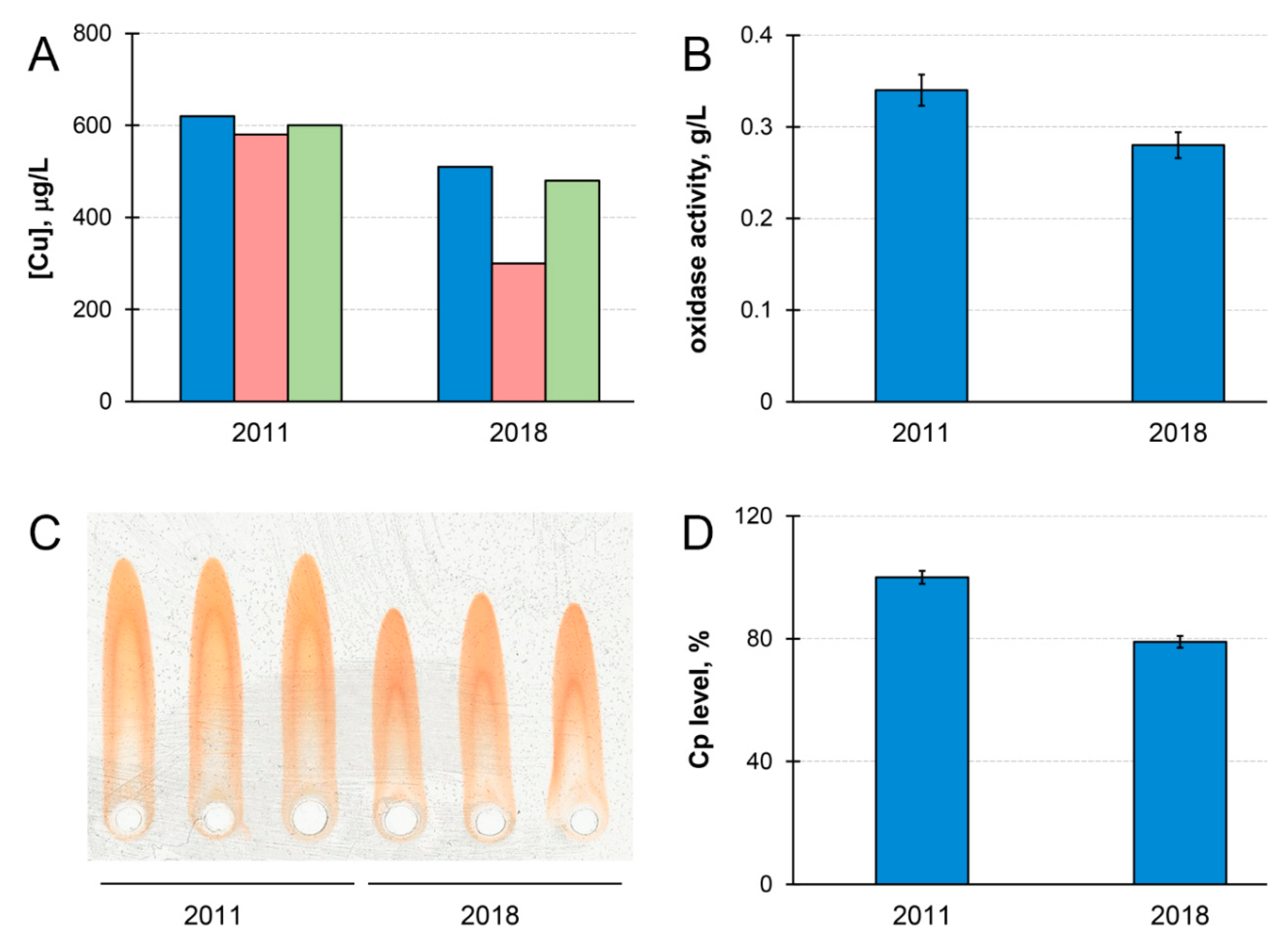
3. Case Report
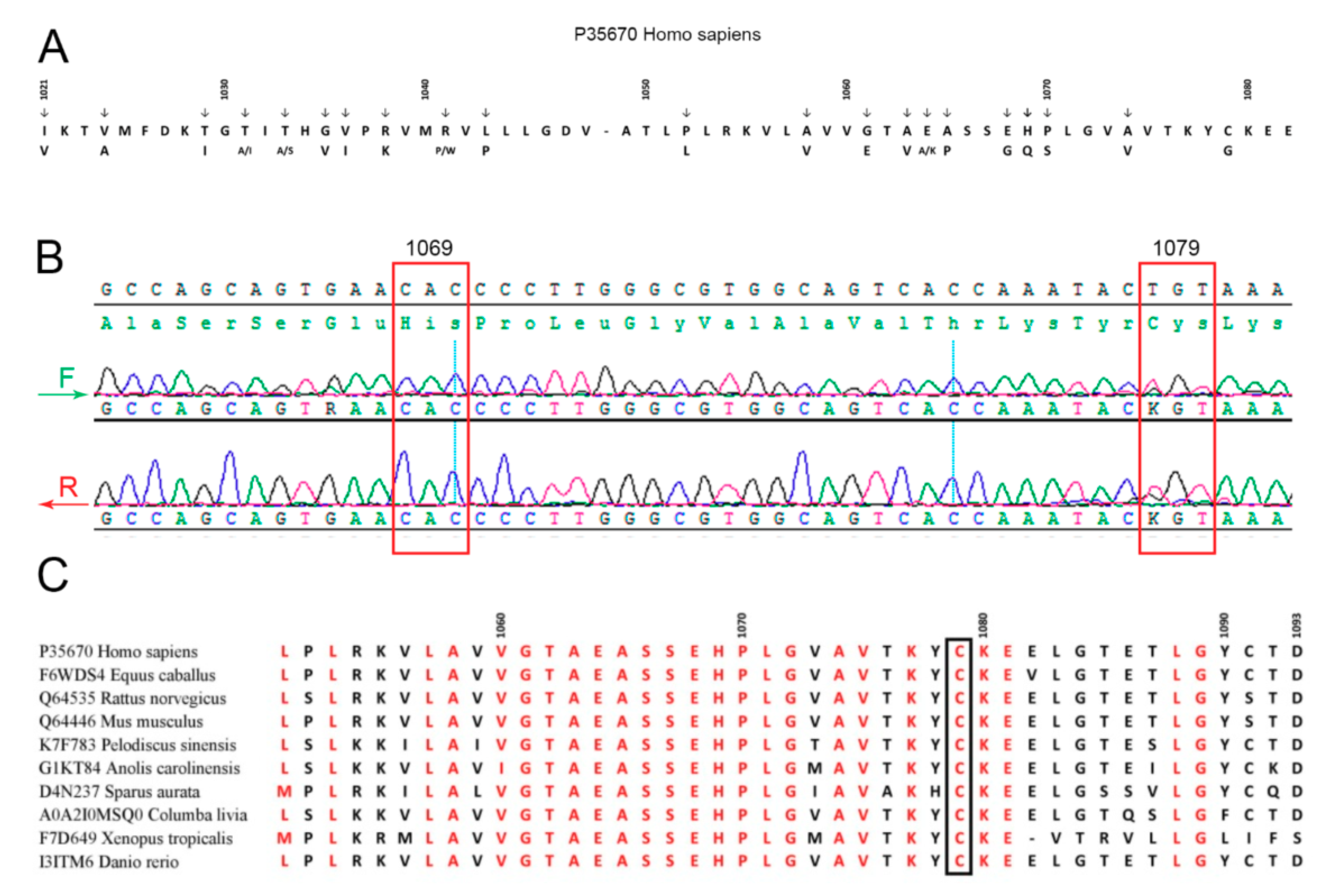
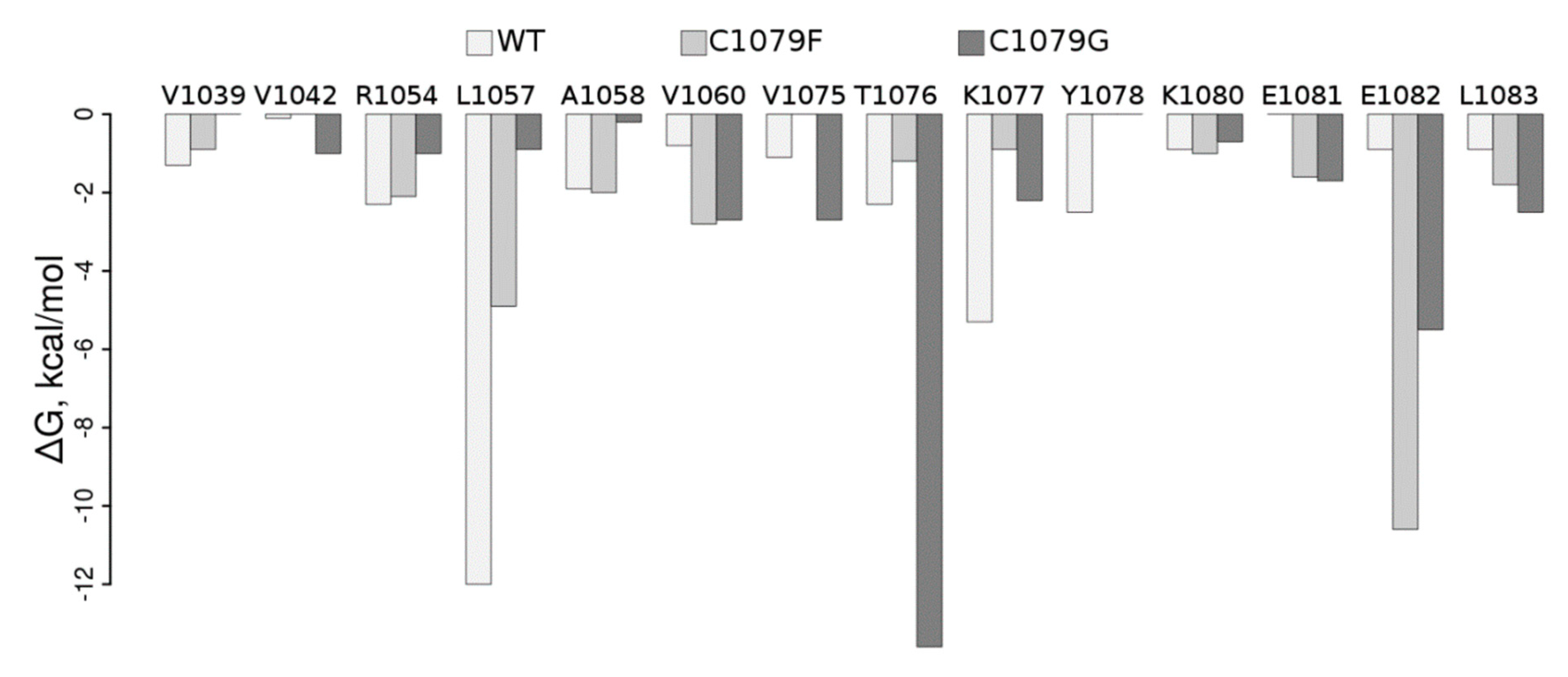
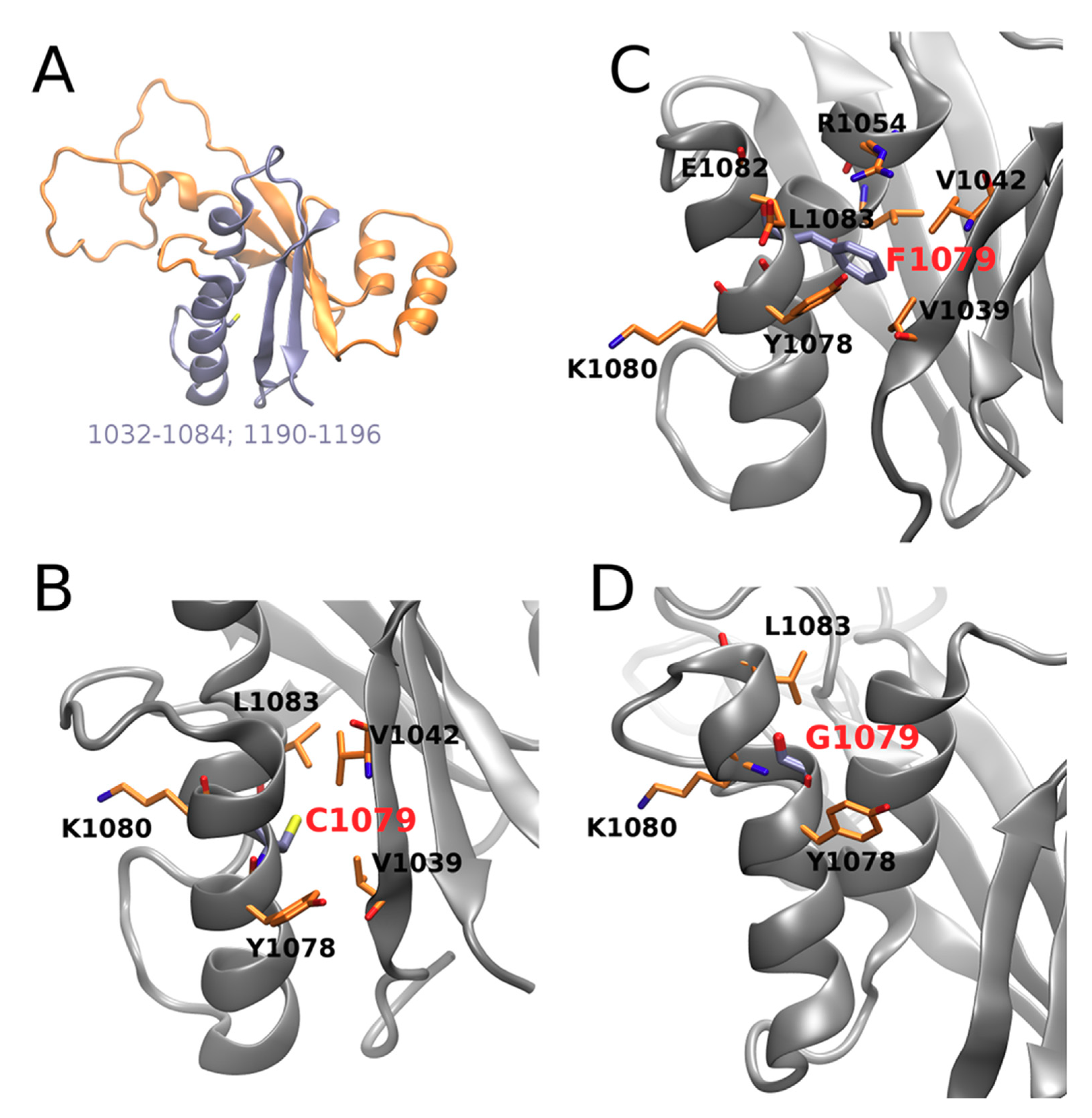
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ATP7B | Cu-transporting ATPase P1 type |
| BDI | Beck’s Depression Inventor |
| Cp | ceruloplasmin |
| CSI | copper status indexes |
| EOPD | early onset Parkinson’s disease |
| FAB | Frontal Assessment Battery |
| HADS | Hospital Anxiety and Depression Scale |
| H&Y | Hoehn and Yahr stage |
| IL | interleukin |
| MD | molecular dynamics |
| MM-GBSA | Molecular Mechanics-Generalized Borne Surface Area |
| MMSE | Mini-Mental State Examination |
| N-domain | nucleotide-binding domain |
| NCC | non-Cp copper |
| NMS PD | Non-Motor Symptoms Scale for Parkinson’s disease |
| PD | Parkinson’s disease |
| pPD | patient with PD |
| PIGD | Postural Instability and Gait Disorder |
| ROS | radical oxygen species |
| SE-ADL | Schwab and England Activities of Daily Living Scale |
| SNpc | substantia nigra pars compacta |
| TNF | tumor necrosis factor |
| UPDRS | Unified Parkinson’s Disease Rating Scale |
| WD | Wilson’s disease |
| WT | wild type |
References
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Sveinbjornsdottir, S. The clinical symptoms of Parkinson’s disease. J. Neurochem. 2016, 139, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.K.Y.; Xu, Y.H.; Chan, L.K.M.; Braidy, N.; Mellick, G.D. Mini-review on initiatives to interfere with the propagation and clearance of alpha-synuclein in Parkinson’s disease. Transl. Neurodegener. 2017, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Al Shahrani, M.; Heales, S.; Hargreaves, I.; Orford, M. Oxidative Stress: Mechanistic Insights into Inherited Mitochondrial Disorders and Parkinson’s Disease. J. Clin. Med. 2017, 6, 100. [Google Scholar] [CrossRef] [PubMed]
- Carboni, E.; Lingor, P. Insights on the interaction of alpha-synuclein and metals in the pathophysiology of Parkinson’s disease. Metallomics 2015, 7, 395–404. [Google Scholar] [CrossRef]
- Dell’Acqua, S.; Pirota, V.; Anzani, C.; Rocco, M.M.; Nicolis, S.; Valensin, D.; Monzani, E.; Casella, L. Reactivity of copper–α-synuclein peptide complexes relevant to Parkinson’s disease. Metallomics 2015, 7, 1091–1102. [Google Scholar] [CrossRef]
- Villar-Pique, A.; Rossetti, G.; Ventura, S.; Carloni, P.; Fernandez, C.O.; Outeiro, T.F. Copper (II) and the pathological H50Q α-synuclein mutant: Environment meets genetics. Commun. Integr. Biol. 2017, 10, e1270484. [Google Scholar] [CrossRef]
- Jin, L.; Wang, J.; Zhao, L.; Jin, H.; Fei, G.; Zhang, Y.; Zeng, M.; Zhong, C. Decreased serum ceruloplasmin levels characteristically aggravate nigral iron deposition in Parkinson’s disease. Brain 2011, 134, 50–58. [Google Scholar] [CrossRef]
- Ilyechova, E.Y.; Miliukhina, I.V.; Orlov, I.A.; Muruzheva, Z.M.; Puchkova, L.V.; Karpenko, M.N. A low blood copper concentration is a co-morbidity burden factor in Parkinson’s disease development. Neurosci. Res. 2018, 135, 54–62. [Google Scholar] [CrossRef]
- Bharucha, K.J.; Friedman, J.K.; Vincent, A.S.; Ross, E.D. Lower serum ceruloplasmin levels correlate with younger age of onset in Parkinson’s disease. J. Neurol. 2008, 255, 1957–1962. [Google Scholar] [CrossRef] [PubMed]
- Bielli, P.; Calabrese, L. Structure to function relationships in ceruloplasmin: A ’moonlighting’ protein. Cell. Mol. Life Sci. 2002, 59, 1413–1427. [Google Scholar] [CrossRef] [PubMed]
- Ramos, D.; Mar, D.; Ishida, M.; Vargas, R.; Gaite, M.; Montgomery, A.; Linder, M.C. Mechanism of Copper Uptake from Blood Plasma Ceruloplasmin by Mammalian Cells. PLoS ONE 2016, 11, e0149516. [Google Scholar] [CrossRef] [PubMed]
- Golenkina, E.A.; Viryasova, G.M.; Galkina, S.I.; Gaponova, T.V.; Sud’Ina, G.F.; Sokolov, A.V. Fine Regulation of Neutrophil Oxidative Status and Apoptosis by Ceruloplasmin and Its Derivatives. Cells 2018, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Sahoo, P.K. Ceruloplasmin, a moonlighting protein in fish. Fish Shellfish Immunol. 2018, 82, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-Y.; Zhuang, Q.-Q.; Zhu, L.-B.; Zhu, H.; Li, T.; Li, R.; Chen, S.-F.; Huang, C.-P.; Zhang, X.; Zhu, J.-H. Meta-analysis of brain iron levels of Parkinson’s disease patients determined by postmortem and MRI measurements. Sci. Rep. 2016, 6, 36669. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Wang, J.; Rogers, J.; Xie, J. Brain iron metabolism dysfunction in Parkinson’s disease. Mol. Neurobiol. 2017, 54, 3078–3101. [Google Scholar] [CrossRef] [PubMed]
- Montes, S.; Rivera-Mancía, S.; Díaz-Ruíz, A.; Tristán-López, L.; Ríos, C. Copper and Copper Proteins in Parkinson’s Disease. Oxid. Med. Cell. Longev. 2014, 2014, 147251. [Google Scholar] [CrossRef]
- Davies, K.M.; Mercer, J.F.; Chen, N.; Double, K.L. Copper dyshomoeostasis in Parkinson’s disease: Implications for pathogenesis and indications for novel therapeutics. Clin. Sci. 2016, 130, 565–574. [Google Scholar] [CrossRef]
- Cruces-Sande, A.; Rodríguez-Pérez, A.I.; Herbello-Hermelo, P.; Bermejo-Barrera, P.; Méndez-Álvarez, E.; Labandeira-García, J.L.; Soto-Otero, R. Copper Increases Brain Oxidative Stress and Enhances the Ability of 6-Hydroxydopamine to Cause Dopaminergic Degeneration in a Rat Model of Parkinson’s Disease. Mol. Neurobiol. 2018, 56, 2845–2854. [Google Scholar] [CrossRef]
- Redenšek, S.; Trost, M.; Dolžan, V. Genetic Determinants of Parkinson’s Disease: Can They Help to Stratify the Patients Based on the Underlying Molecular Defect? Front. Aging Neurosci. 2017, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Wang, P.; Jankovic, J. The genetics of Parkinson disease. Ageing Res. Rev. 2018, 42, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.-S.; Geng, W.-S.; Jia, J.-J.; Chen, L.; Zhang, P.-P. Cellular and Molecular Basis of Neurodegeneration in Parkinson Disease. Front. Aging Neurosci. 2018, 10, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimi-Moghadam, A.; Charsouei, S.; Bell, B.; Jabalameli, M.R. Parkinson Disease from Mendelian Forms to Genetic Susceptibility: New Molecular Insights into the Neurodegeneration Process. Cell. Mol. Neurobiol. 2018, 38, 1153–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramesh, J.; Ronsard, L.; Gao, A.; Venugopal, B. Autophagy Intertwines with Different Diseases—Recent Strategies for Therapeutic Approaches. Diseases 2019, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Torrent, R.; Rigotti, F.D.A.; Dell’Era, P.; Memo, M.; Raya, A.; Consiglio, A. Using iPS Cells toward the Understanding of Parkinson’s Disease. J. Clin. Med. 2015, 4, 548–566. [Google Scholar] [CrossRef] [PubMed]
- Wirdefeldt, K.; Adami, H.-O.; Cole, P.; Trichopoulos, D.; Mandel, J. Epidemiology and etiology of Parkinson’s disease: A review of the evidence. Eur. J. Epidemiol. 2011, 26, 1–58. [Google Scholar] [CrossRef]
- Hochstrasser, H.; Bauer, P.; Walter, U.; Behnke, S.; Spiegel, J.; Csoti, I.; Zeiler, B.; Bornemann, A.; Pahnke, J.; Becker, G.; et al. Ceruloplasmin gene variations and substantia nigra hyperechogenicity in Parkinson disease. Neurology 2004, 63, 1912–1917. [Google Scholar] [CrossRef]
- Zhao, N.; Jin, L.; Fei, G.; Zheng, Z.; Zhong, C. Serum microRNA-133b is associated with low ceruloplasmin levels in Parkinson’s disease. Park. Relat. Disord. 2014, 20, 1177–1180. [Google Scholar] [CrossRef]
- Limphaibool, N.; Iwanowski, P.; Holstad, M.J.V.; Perkowska, K. Parkinsonism in Inherited Metabolic Disorders: Key Considerations and Major Features. Front. Neurol. 2018, 9, 857. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Wang, X.P. Does ceruloplasmin defend against neurodegenerative diseases? Curr. Neuropharmacol. 2019, 17, 539–549. [Google Scholar] [CrossRef]
- Emelyanov, A.K.; Usenko, T.S.; Tesson, C.; Senkevich, K.A.; Nikolaev, M.A.; Miliukhina, I.V.; Kopytova, A.E.; Timofeeva, A.A.; Yakimovsky, A.F.; Lesage, S.; et al. Mutation analysis of Parkinson’s disease genes in a Russian data set. Neurobiol. Aging 2018, 71, 267.e7–267.e10. [Google Scholar] [CrossRef]
- Lutsenko, S.; Chernov, I.; Ross, B.M.; Gilliam, T.; Petrukhin, K.; Kaplan, J.H. Characterization of the Wilson disease gene encoding a P-type copper transporting ATPase: Genomic organization, alternative splicing, and structure/function predictions. Hum. Mol. Genet. 1994, 3, 1647–1656. [Google Scholar]
- Schilsky, M.L. Wilson disease: Diagnosis, treatment, and follow-up. Clin. Liver Dis. 2017, 21, 755–767. [Google Scholar] [CrossRef]
- Polishchuk, R.; Lutsenko, S. GOLGI IN COPPER HOMEOSTASIS: A view from the membrane trafficking field. Histochem. Cell Biol. 2013, 140, 285–295. [Google Scholar] [CrossRef]
- Högl, B.; Arnulf, I.; Comella, C.; Ferreira, J.; Iranzo, A.; Tilley, B.; et al. Scales to assess sleep impairment in Parkinson’s disease: Critique and recommendations. Movement Disorders 2010, 25, 2704–2716. [Google Scholar] [CrossRef]
- Hoehn, M.M.; Yahr, M.D. Parkinsonism: Onset, progression, and mortality. Neurology 1967, 17, 427. [Google Scholar] [CrossRef]
- Chaudhuri, K.R.; Rojo, J.M.; Schapira, A.H.V.; Brooks, D.J.; Stocchi, F.; Odin, P.; Antonini, A.; Brown, R.J.; Martinez-Martin, P. A Proposal for a Comprehensive Grading of Parkinson’s Disease Severity Combining Motor and Non-Motor Assessments: Meeting an Unmet Need. PLoS ONE 2013, 8, e57221. [Google Scholar] [CrossRef]
- Folstein, M.F.; Folstein, S.E.; McHugh, P.R. Mini-mental state. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 1975, 12, 189–198. [Google Scholar] [CrossRef]
- Beck, A.T.; Ward, C.H.; Mendelson, M.; Mock, J.; Erbaugh, J. An Inventory for Measuring Depression. Arch. Gen. Psychiatry 1961, 4, 561. [Google Scholar] [CrossRef]
- Snaith, R.P.; Zigmond, A.S. The hospital anxiety and depression scale. Br. Med. J. (Clin. Res. Ed.) 1986, 292, 344. [Google Scholar] [CrossRef]
- Dubois, B.; Slachevsky, A.; Litvan, I.; Pillon, B. The FAB: A frontal assessment battery at bedside. Neurology 2000, 55, 1621–1626. [Google Scholar] [CrossRef] [Green Version]
- Dagur, P.K.; McCoy, J.P. Collection, storage, and preparation of human blood cells. Curr. Protoc. Cytom. 2016, 73, 5.1.1–5.1.16. [Google Scholar] [CrossRef]
- Thomas, G.R.; Forbes, J.R.; Roberts, E.A.; Walshe, J.M.; Cox, D.W. The Wilson disease gene: Spectrum of mutations and their consequences. Nat. Genet. 1995, 9, 210–217. [Google Scholar] [CrossRef]
- Sunderman, F.W., Jr.; Nomoto, S. Measurement of human serum ceruloplasmin by its p-phenylenediamine oxidase activity. Clin. Chem. 1970, 16, 903–910. [Google Scholar]
- Zatulovskaia, Y.A.; Ilyechova, E.Y.; Puchkova, L.V. The Features of Copper Metabolism in the Rat Liver during Development. PLoS ONE 2015, 10, e0140797. [Google Scholar] [CrossRef]
- Sokolov, A.V.; Kostevich, V.A.; Romanico, D.N.; Zakharova, E.T.; Vasilyev, V.B.; Sokolov, A. Two-stage method for purification of ceruloplasmin based on its interaction with neomycin. Biochemistry (Moscow) 2012, 77, 631–638. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K. AMBER16; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Dmitriev, O.; Tsivkovskii, R.; Abildgaard, F.; Morgan, C.T.; Markley, J.L.; Lutsenko, S. Solution structure of the N-domain of Wilson disease protein: Distinct nucleotide-binding environment and effects of disease mutations. Proc. Natl. Acad. Sci. USA 2006, 103, 5302–5307. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Tsui, V.; Case, D.A. Theory and applications of the generalized born solvation model in macromolecular simulations. Biopolymers 2000, 56, 275–291. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Modification of the Generalized Born Model Suitable for Macromolecules. J. Phys. Chem. B 2000, 104, 3712–3720. [Google Scholar] [CrossRef] [Green Version]
- Homeyer, N.; Gohlke, H. Free Energy Calculations by the Molecular Mechanics Poisson—Boltzmann Surface Area Method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Gupta, V.; Garg, R.K.; Khattri, S. Levels of IL-8 and TNF-α decrease in Parkinson’s disease. Neurol. Res. 2016, 38, 1–5. [Google Scholar] [CrossRef]
- Kalita, J.; Kumar, V.; Misra, U.K. A study on apoptosis and anti-apoptotic status in Wilson disease. Mol. Neurobiol. 2016, 53, 6659–6667. [Google Scholar] [CrossRef]
- Dursun, E.; Gezen-Ak, D.; Hanağası, H.; Bilgic, B.; Lohmann, E.; Ertan, S.; Atasoy, I.L.; Alaylıoğlu, M.; Araz Ömür, S.; Onal, B.; et al. The interleukin 1 alpha, interleukin 1 beta, interleukin 6 and alpha-2-macroglobulin serum levels in patients with early or late onset Alzheimer’s disease, mild cognitive impairment or Parkinson’s disease. J. Neuroimmunol. 2015, 283, 50–57. [Google Scholar] [CrossRef]
- Goyal, M.K.; Sinha, S.; Patil, S.A.; Jayalekshmy, V.; Taly, A.B. Do cytokines have any role in Wilson’s disease? Clin. Exp. Immunol. 2008, 154, 74–79. [Google Scholar] [CrossRef]
- Bernevic, B.; El-Khatib, A.H.; Jakubowski, N.; Weller, M.G. Online immunocapture ICP-MS for the determination of the metalloprotein ceruloplasmin in human serum. BMC Res. Notes 2018, 11, 213. [Google Scholar] [CrossRef]
- Woimant, F.; Djebrani-Oussedik, N.; Poujois, A. New tools for Wilson’s disease diagnosis: Exchangeable copper fraction. Ann. Transl. Med. 2019, 7, S70. [Google Scholar] [CrossRef]
- Zhang, Q.J.; Xu, L.Q.; Wang, C.; Hu, W.; Wang, N.; Chen, W.J. Four-year follow-up of a Wilson disease pedigree complicated with epilepsy and hypopituitarism: Case report with a literature review. Medicine (Baltimore) 2016, 95, e5331. [Google Scholar] [CrossRef]
- Gray, L.W.; Peng, F.; Molloy, S.A.; Pendyala, V.S.; Muchenditsi, A.; Muzik, O.; Lee, J.; Kaplan, J.H.; Lutsenko, S. Urinary Copper Elevation in a Mouse Model of Wilson’s Disease Is a Regulated Process to Specifically Decrease the Hepatic Copper Load. PLoS ONE 2012, 7, e38327. [Google Scholar] [CrossRef]
- Shah, A.B.; Chernov, I.; Zhang, H.T.; Ross, B.M.; Das, K.; Lutsenko, S.; Parano, E.; Pavone, L.; Evgrafov, O.; Ivanova-Smolenskaya, I.A.; et al. Identification and analysis of mutations in the Wilson disease gene (ATP7B): Population frequencies, genotype-phenotype correlation, and functional analyses. Am. J. Hum. Genet. 1997, 61, 317–328. [Google Scholar] [CrossRef]
- Ivanova-Smolenska, I.A.; Ovchinnikov, I.V.; Karabanov, A.V.; Deineko, N.L.; Poleshchuk, V.V.; Markova, E.D.; Illarioshkin, S.N. The His1069Gln mutation in the ATP7B gene in Russian patients with Wilson disease. J. Med. Genet. 1999, 36, 174. [Google Scholar]
- Wang, L.-H.; Huang, Y.-Q.; Shang, X.; Su, Q.-X.; Xiong, F.; Yu, Q.-Y.; Lin, H.-P.; Wei, Z.-S.; Hong, M.-F.; Xu, X.-M. Mutation analysis of 73 southern Chinese Wilson’s disease patients: Identification of 10 novel mutations and its clinical correlation. J. Hum. Genet. 2011, 56, 660–665. [Google Scholar] [CrossRef]
- Chou, P.Y.; Fasman, G.D. Prediction of protein conformation. Biochemistry 1974, 13, 222–245. [Google Scholar] [CrossRef]
- Eyal, E.; Lum, G.; Bahar, I. The anisotropic network model web server at 2015 (ANM 2.0). Bioinformatics 2015, 31, 1487–1489. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Ralle, M.; Huster, D.; Vogt, S.; Schirrmeister, W.; Burkhead, J.L.; Capps, T.R.; Gray, L.; Lai, B.; Maryon, E.; Lutsenko, S.; et al. Wilson disease at a single cell level: Intracellular copper trafficking activates compartment-specific responses in hepatocytes. J. Biol. Chem. 2010, 285, 30875–30883. [Google Scholar] [CrossRef]
- Lichtmannegger, J.; Leitzinger, C.; Wimmer, R.; Schmitt, S.; Schulz, S.; Kabiri, Y.; Eberhagen, C.; Rieder, T.; Janik, D.; Neff, F.; et al. Methanobactin reverses acute liver failure in a rat model of Wilson disease. J. Clin. Investig. 2016, 126, 2721–2735. [Google Scholar] [CrossRef]
- Okita, Y.; Rcom-H’cheo-Gauthier, A.N.; Goulding, M.; Chung, R.S.; Faller, P.; Pountney, D.L. Metallothionein, copper and alpha-synuclein in alpha-synucleinopathies. Front. Neurosci. 2017, 11, 114. [Google Scholar] [CrossRef]
- Bento, I.; Peixoto, C.; Zaitsev, V.N.; Lindley, P.F. Ceruloplasmin revisited: Structural and functional roles of various metal cation-binding sites. Acta Crystallogr. Sect. D Biol. Crystallogr. 2007, 63, 240–248. [Google Scholar] [CrossRef]
- Fukushima, T.; Tan, X.; Luo, Y.; Wang, P.; Song, J.; Kanda, H.; Hayakawa, T.; Kumagai, T.; Kakamu, T.; Tsuji, M.; et al. Heavy metals in blood and urine and its relation to depressive symptoms in Parkinson’s disease patients. Fukushima J. Med. Sci. 2013, 59, 76–80. [Google Scholar] [CrossRef]
- Kim, M.-J.; Oh, S.-B.; Kim, J.; Kim, K.; Ryu, H.-S.; Kim, M.S.; Ayton, S.; Bush, A.I.; Lee, J.-Y.; Chung, S.J. Association of metals with the risk and clinical characteristics of Parkinson’s disease. Park. Relat. Disord. 2018, 55, 117–121. [Google Scholar] [CrossRef]
- Tsivkovskii, R.; Efremov, R.G.; Lutsenko, S. The Role of the Invariant His-1069 in Folding and Function of the Wilson’s Disease Protein, the Human Copper-transporting ATPase ATP7B. J. Biol. Chem. 2003, 278, 13302–13308. [Google Scholar] [CrossRef]
- Morgan, C.T.; Tsivkovskii, R.; Kosinsky, Y.A.; Efremov, R.G.; Lutsenko, S. The Distinct Functional Properties of the Nucleotide-binding Domain of ATP7B, the Human Copper-transporting ATPase: Analysis of the wilson disease mutations E1064A, H1069Q, R1151H, AND C1104F. J. Biol. Chem. 2004, 279, 36363–36371. [Google Scholar] [CrossRef]
- Johnson, S. Is Parkinson’s disease the heterozygote form of Wilson’s disease: PD = ½ WD? Med. Hypotheses. 2001, 56, 171–173. [Google Scholar] [CrossRef]
- Sechi, G.; Cocco, G.A.; Errigo, A.; Deiana, L.; Rosati, G.; Agnetti, V.; Paulus, K.S.; Pes, G.M. Three sisters with very-late-onset major depression and parkinsonism. Park. Relat. Disord. 2007, 13, 122–125. [Google Scholar] [CrossRef]
- Loudianos, G.; Dessi, V.; Lovicu, M.; Angius, A.; Figus, A.; Lilliu, F.; De Virgiliis, S.; Nurchi, A.M.; Deplano, A.; Moi, P.; et al. Molecular characterization of Wilson disease in the Sardinian population? Evidence of a founder effect. Hum. Mutat. 1999, 14, 294–303. [Google Scholar] [CrossRef]
- Ferenci, P. Regional distribution of mutations of the ATP7B gene in patients with Wilson disease: Impact on genetic testing. Qual. Life Res. 2006, 120, 151–159. [Google Scholar] [CrossRef]
- Möller, J.C.; Leinweber, B.; Rissling, I.; Oertel, W.H.; Bandmann, O.; Schmidt, H.H.-J. Prevalence of the H1069Q mutation inATP7B in discordant pairs with early-onset Parkinson’s Disease. Mov. Disord. 2006, 21, 1789–1790. [Google Scholar] [CrossRef]
- Ala, A.; Borjigin, J.; Rochwarger, A.; Schilsky, M. Wilson disease in septuagenarian siblings: Raising the bar for diagnosis. Hepatology 2005, 41, 668–670. [Google Scholar] [CrossRef]
- Karpenko, M.N.; Ilyicheva, E.Y.; Muruzheva, Z.M.; Milyukhina, I.V.; Orlov, Y.A.; Puchkova, L. Role of Copper Dyshomeostasis in the Pathogenesis of Parkinson’s Disease. Bull. Exp. Biol. Med. 2018, 164, 596–600. [Google Scholar] [CrossRef]
- Członkowska, A.; Litwin, T.; Dusek, P.; Ferenci, P.; Lutsenko, S.; Medici, V.; Rybakowski, J.K.; Weiss, K.H.; Schilsky, M.L. Wilson disease. Nat. Rev. Dis. Primers 2018, 4, 21. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Date of Observation | ||
|---|---|---|---|
| September 2011 | October 2014 | June 2018 | |
| Age (years) | 51 | 54 | 57 |
| Modified H&Y scale (stage) | 1.5 | 2.5 | 2.5 |
| UPDRS I (scores) | 3 | 5 | 6 |
| UPDRS II (scores) | 7 | 10 | 11 |
| UPDRS III (scores) | 24 | 38 | 48 |
| UPDRS IV (scores) | 1 | 5 | 7 |
| Total UPDRS (scores) | 35 | 58 | 72 |
| MMSE (scores) | 30 | 28 | 28 |
| FAB (scores) | 18 | 18 | 18 |
| Clock-drawing test (scores) | 10 | 10 | 10 |
| PD-NMS (scores) | 13 | 26 | 32 |
| BDI’s scale (scores) | 15 | 14 | 17 |
| Sheehan Clinical Anxiety Rating Scale (scores) | 21 | 10 | 35 |
| HADS «A» (scores) | 5 | 4 | 5 |
| HADS «D» (scores) | 7 | 4 | 6 |
| Parameters (pg/mL) | Patient M. | |
|---|---|---|
| September 2011 | June 2018 | |
| Interleukin-1β | 6.4 | 0 |
| TNF-α | 0 | 2 |
| Interleukin-6 | 1.4 | 0 |
| Interleukin-10 | 6 | 3 |
| Interleukin-8 | Not measured | 14 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ilyechova, E.Y.; Miliukhina, I.V.; Karpenko, M.N.; Orlov, I.A.; Puchkova, L.V.; Samsonov, S.A. Case of Early-Onset Parkinson’s Disease in a Heterozygous Mutation Carrier of the ATP7B Gene. J. Pers. Med. 2019, 9, 41. https://doi.org/10.3390/jpm9030041
Ilyechova EY, Miliukhina IV, Karpenko MN, Orlov IA, Puchkova LV, Samsonov SA. Case of Early-Onset Parkinson’s Disease in a Heterozygous Mutation Carrier of the ATP7B Gene. Journal of Personalized Medicine. 2019; 9(3):41. https://doi.org/10.3390/jpm9030041
Chicago/Turabian StyleIlyechova, Ekaterina Y., Irina V. Miliukhina, Marina N. Karpenko, Iurii A. Orlov, Ludmila V. Puchkova, and Sergey A. Samsonov. 2019. "Case of Early-Onset Parkinson’s Disease in a Heterozygous Mutation Carrier of the ATP7B Gene" Journal of Personalized Medicine 9, no. 3: 41. https://doi.org/10.3390/jpm9030041