The Transcriptomic Toolbox: Resources for Interpreting Large Gene Expression Data within a Precision Medicine Context for Metabolic Disease Atherosclerosis

 ,
,
Abstract
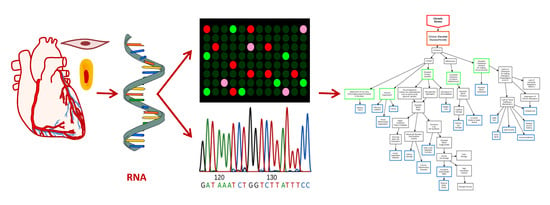
:
1. Introduction
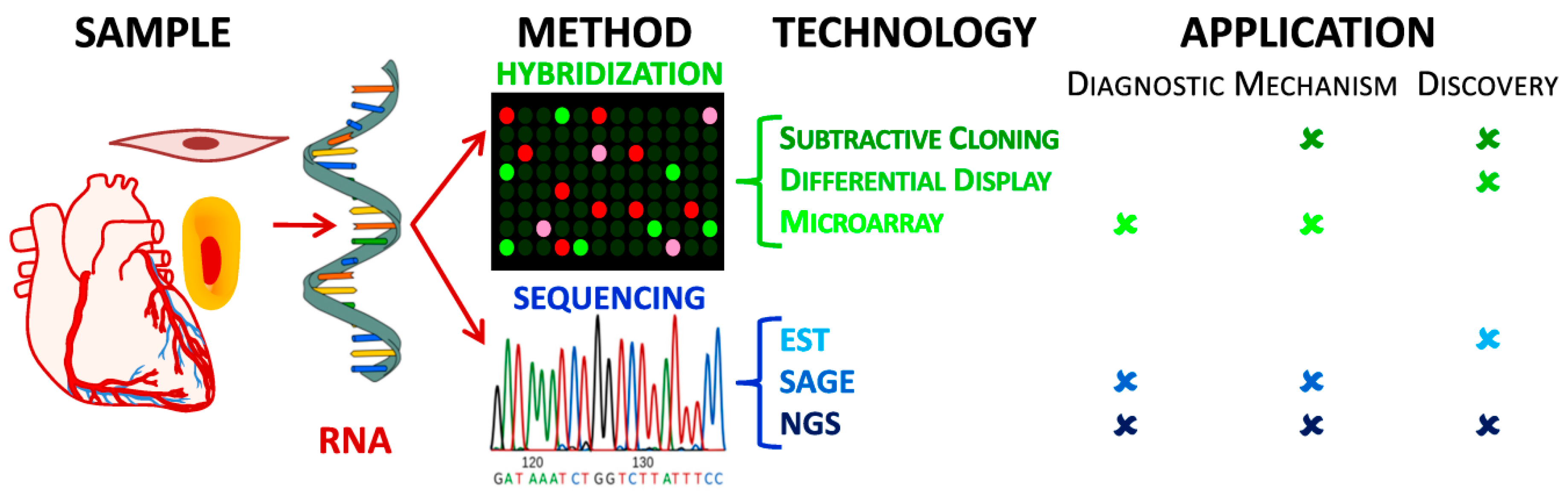
2. Materials and Methods Used in Transcriptomic Studies
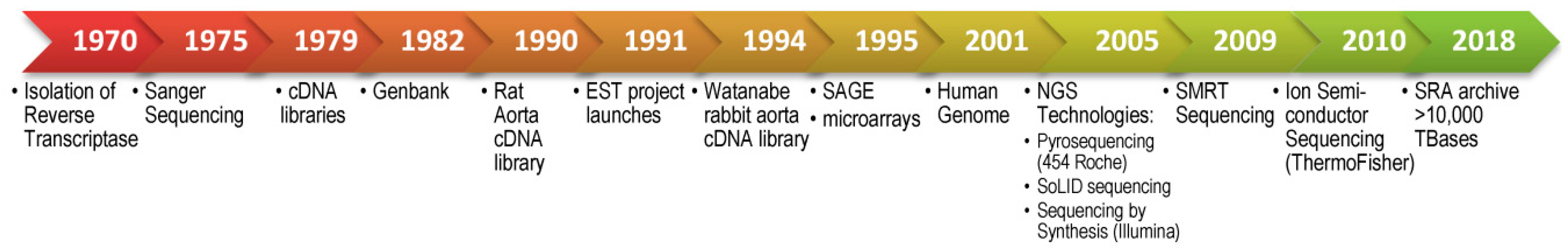
2.1. Origins of Transcriptomics: Gene Expression in the Evolution of Hybridization Techniques and Sequencing for RNA Identification
2.1.1. Classic Hybridization-Based Technologies: Subtractive Cloning & Differential Display
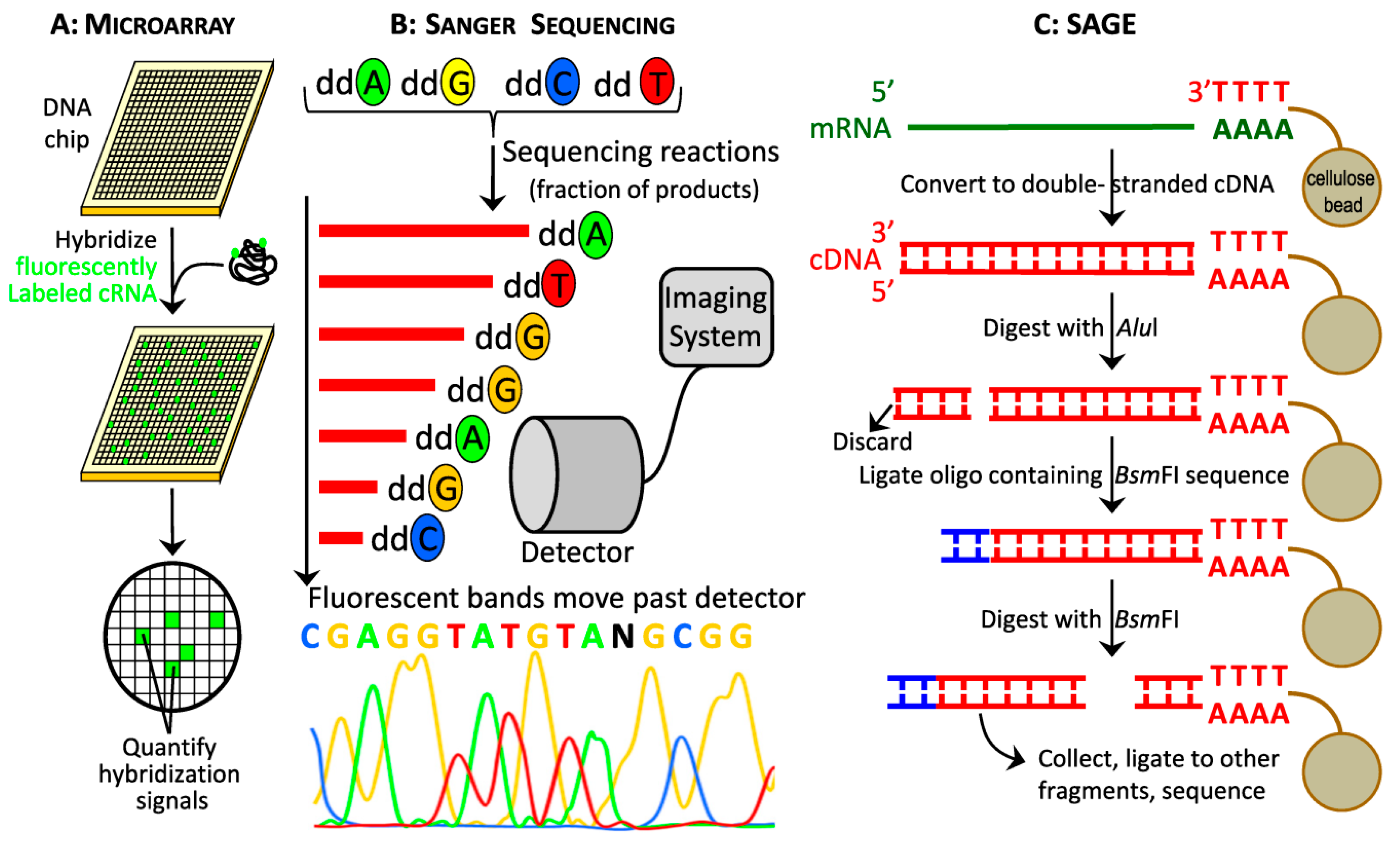
2.1.2. Modern Hybridization-Based Technologies: Microarrays
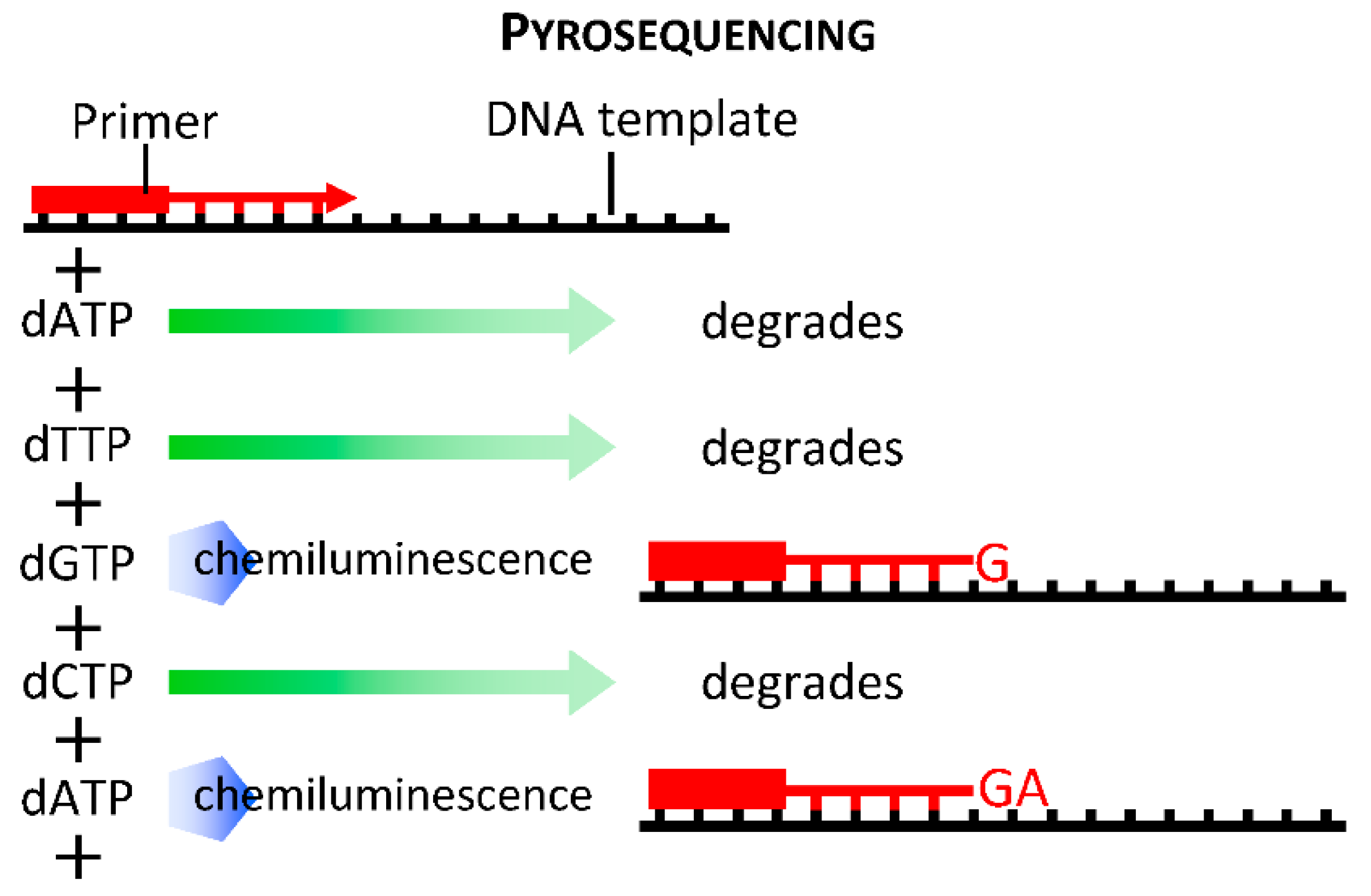
2.1.3. First Generation Sequencing: Sanger Sequencing, cDNA libraries, EST and SAGE
2.2. Next-Generation Sequencing and Deep Transcriptome Analysis
2.2.1. Basic Principles of NGS Sequencing
2.2.2. Development of Single-Cell RNA Sequencing Strategies
2.2.3. Strengths & Caveats for Transcriptome Analysis
2.3. Third Generation Sequencing
Strengths & Caveats for Transcriptome Analysis
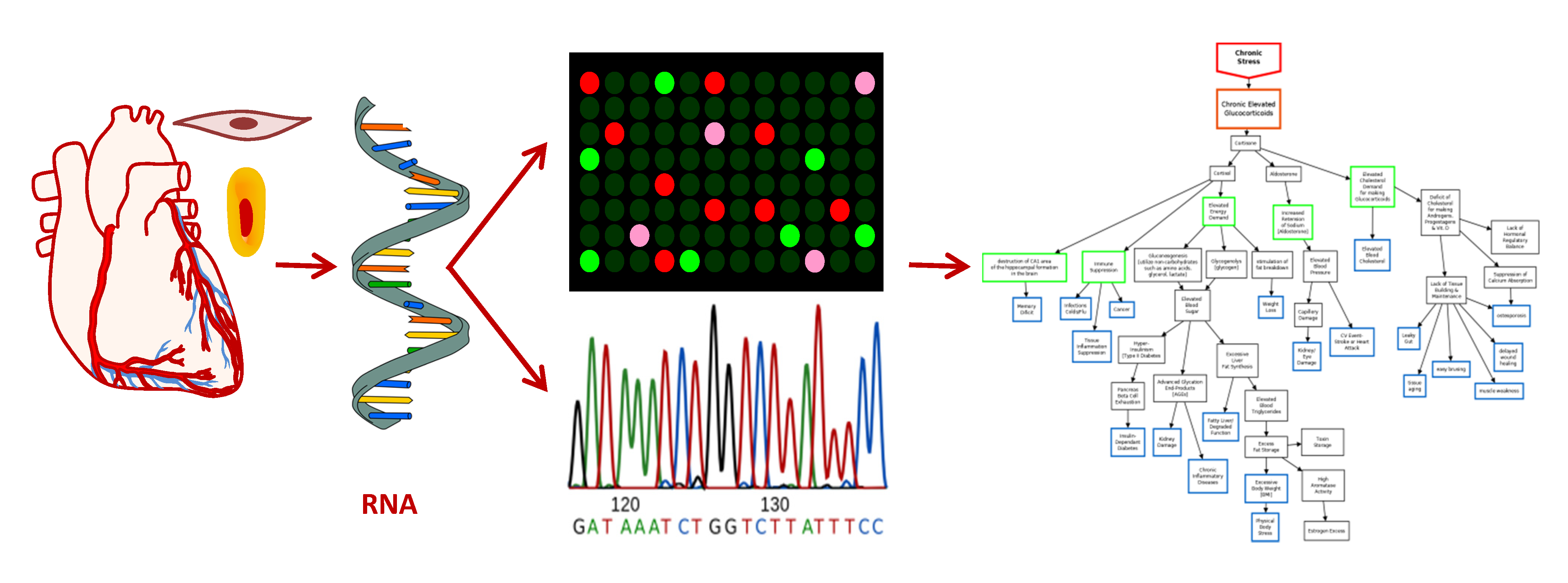
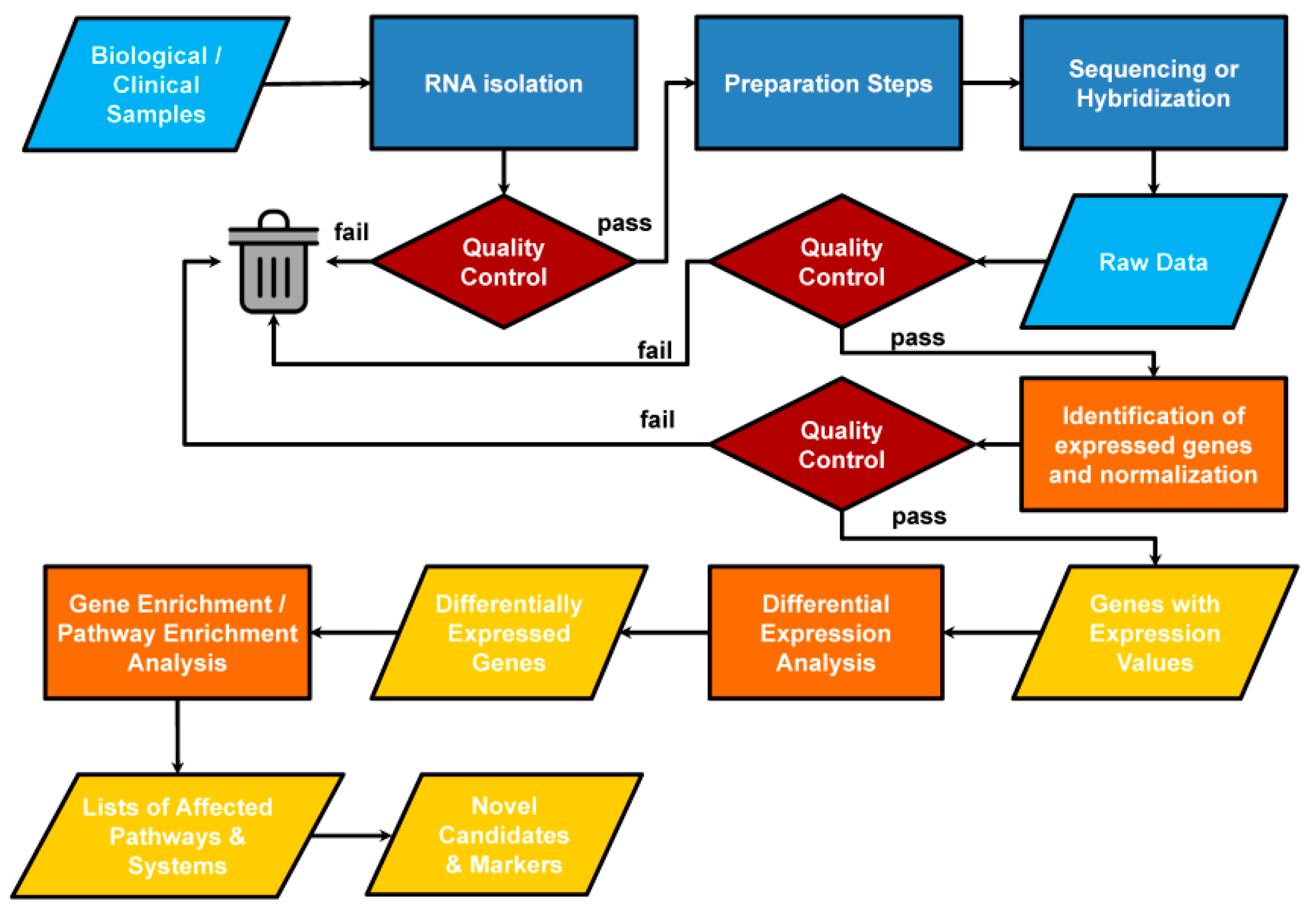
3. Results of Transcriptome Analysis: Unbiased Data Mining to Find a Needle in a Haystack
3.1. Differential Expression Analysis
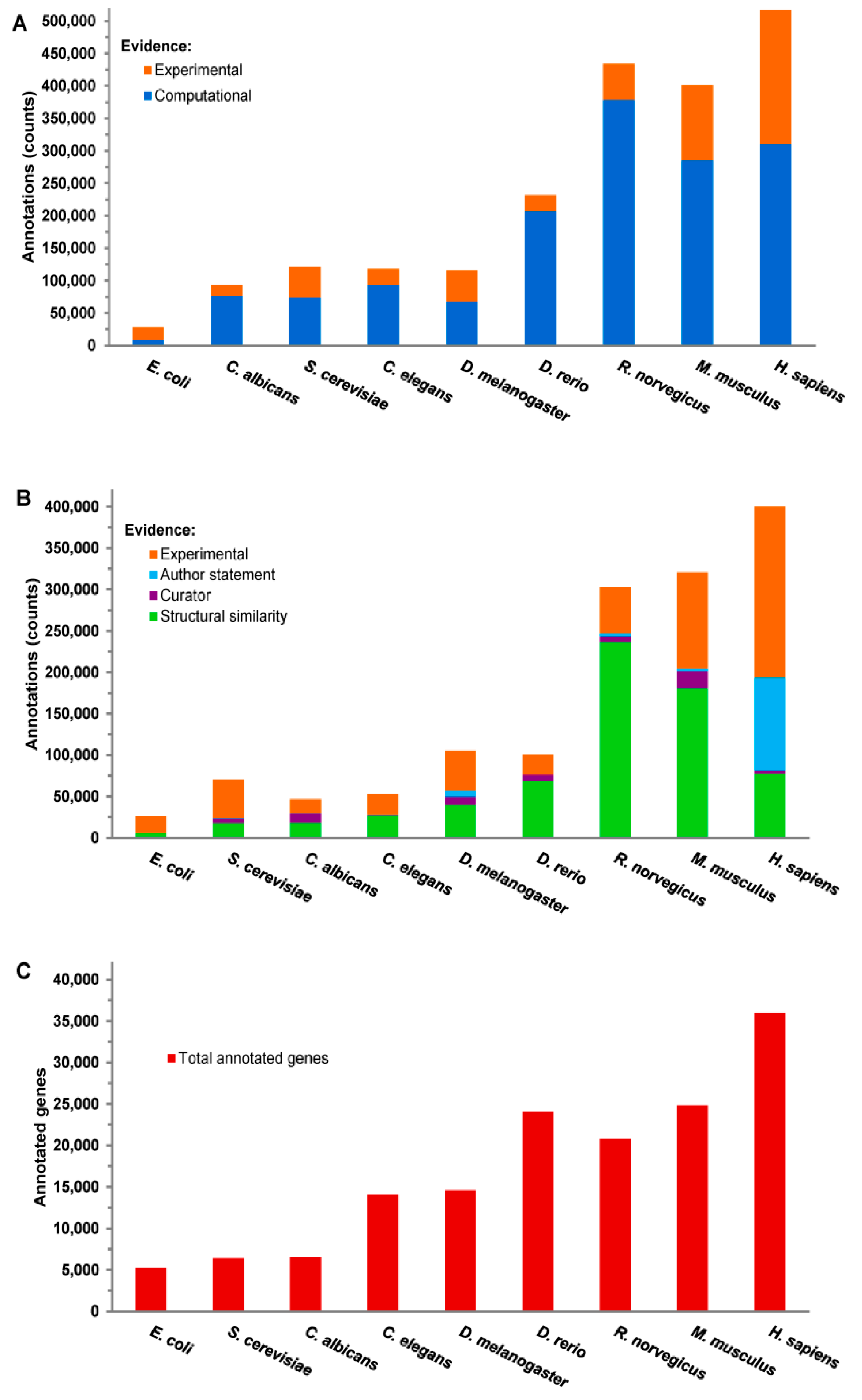
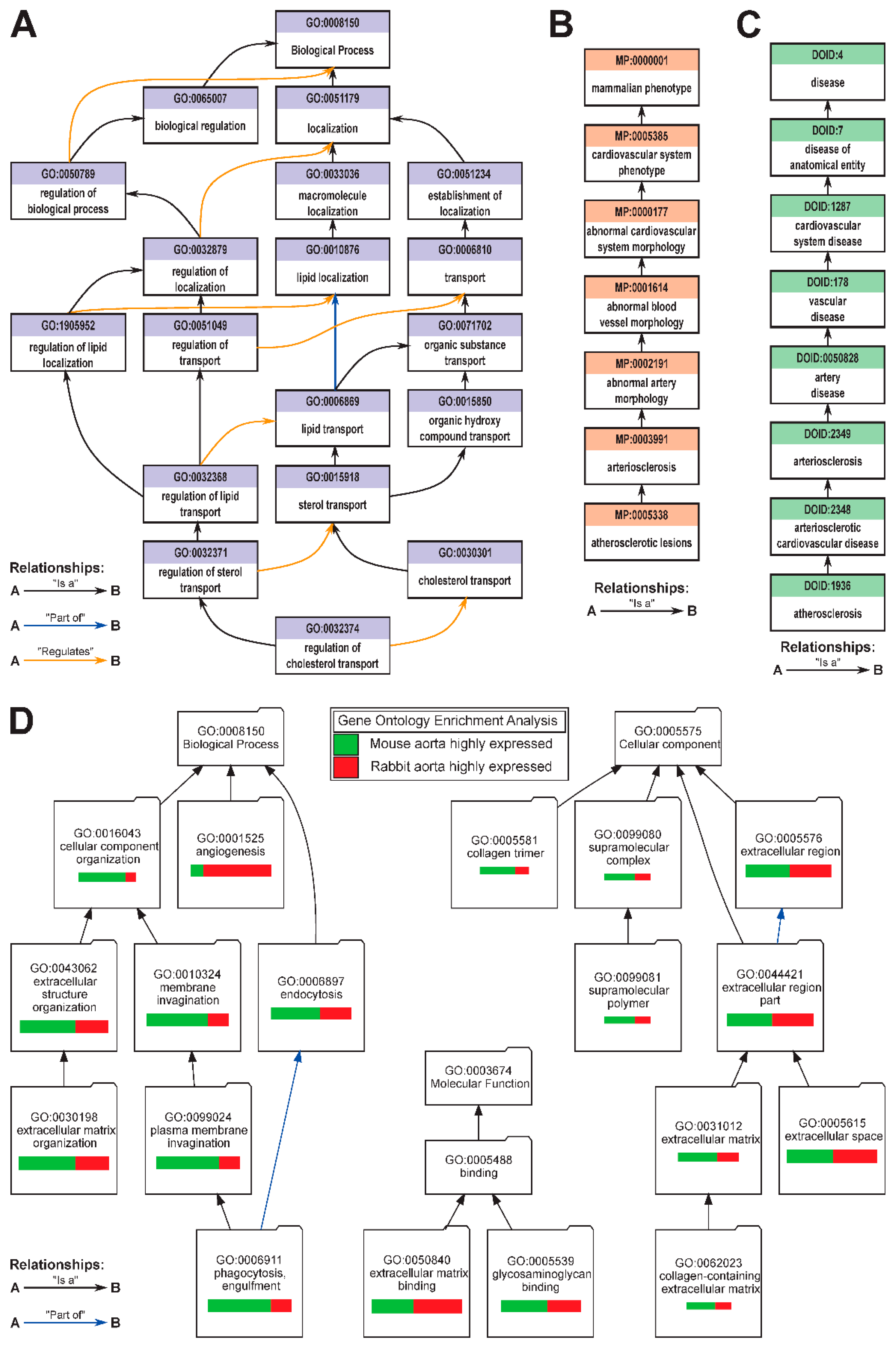
3.2. Categorical Enrichment Analysis: An Overview of Biological Ontologies
4. Discussion: Using Ontologies & Pathway Analysis for Precision Medicine
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McNeal, C.J.; Dajani, T.; Wilson, D.; Cassidy-Bushrow, A.E.; Dickerson, J.B.; Ory, M. Hypercholesterolemia in youth: Opportunities and obstacles to prevent premature atherosclerotic cardiovascular disease. Curr. Atheroscler. Rep. 2010, 12, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Tresch, D.D.; Aronow, W.S. Tresch and Aronow’s Cardiovascular Disease in the Elderly, 5th ed.; CRC Press: Boca Raton, FL, USA, 2014; p. 800. [Google Scholar]
- Ohsfeldt, R.L.; Gandhi, S.K.; Fox, K.M.; Bullano, M.F.; Davidson, M. Medical and cost burden of atherosclerosis among patients treated in routine clinical practice. J. Med. Econ. 2010, 13, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Kochanek, K.D.; Murphy, S.L.; Xu, J.Q.; Arias, E. Mortality in the United States, 2013; National Center for Health Statistics: Hyattsville, MD, USA, 2014.
- Torio, C.M.; Moore, B.J. National Inpatient Hospital Costs: The Most Expensive Conditions by Payer, 2013; Agency for Healthcare Research and Quality: Rockville, MD, USA, 2016.
- Pant, S.; Deshmukh, A.; GuruMurthy, G.S.; Pothineni, N.V.; Watts, T.E.; Romeo, F.; Mehta, J.L. Inflammation and atherosclerosis—Revisited. J. Cardiovasc. Pharmacol. Ther. 2014, 19, 170–178. [Google Scholar] [CrossRef]
- Lau, W.L.; Ix, J.H. Clinical detection, risk factors, and cardiovascular consequences of medial arterial calcification: A pattern of vascular injury associated with aberrant mineral metabolism. Semin. Nephrol. 2013, 33, 93–105. [Google Scholar] [CrossRef] [PubMed]
- McGill, H.C.; McMahan, C.A.; Gidding, S.S. Preventing heart disease in the 21st century. Circulation 2008, 117, 1216–1227. [Google Scholar] [CrossRef] [PubMed]
- Torres, N.; Guevara-Cruz, M.; Velázquez-Villegas, L.A.; Tovar, A.R. Nutrition and atherosclerosis. Arch. Med. Res. 2015, 46, 408–426. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef]
- O’Malley, J.P.; Maslen, C.L.; Illingworth, D.R. Angiotensin-converting enzyme DD genotype and cardiovascular disease in heterozygous familial hypercholesterolemia. Circulation 1998, 97, 1780–1783. [Google Scholar] [CrossRef]
- Huang, S.; Shui, X.; He, Y.; Xue, Y.; Li, J.; Li, G.; Lei, W.; Chen, C. AhR expression and polymorphisms are associated with risk of coronary arterial disease in Chinese population. Sci. Rep. 2015, 5, 8022. [Google Scholar] [CrossRef]
- Slooter, A.J.; van Duijn, C.M.; Bots, M.L.; Ott, A.; Breteler, M.B.; De Voecht, J.; Wehnert, A.; de Knijff, P.; Havekes, L.M.; Grobbee, D.E.; et al. Apolipoprotein e genotype, atherosclerosis, and cognitive decline: The Rotterdam study. J. Neural Transm. Suppl. 1998, 53, 17–29. [Google Scholar]
- Elosua, R.; Ordovas, J.M.; Cupples, L.A.; Fox, C.S.; Polak, J.F.; Wolf, P.A.; D’Agostino, R.A.; O’Donnell, C.J. Association of apoe genotype with carotid atherosclerosis in men and women: The framingham heart study. J. Lipid Res. 2004, 45, 1868–1875. [Google Scholar] [CrossRef] [PubMed]
- Sagerström, C.G.; Sun, B.I.; Sive, H.L. Subtractive cloning: Past, present, and future. Annu. Rev. Biochem. 1997, 66, 751–783. [Google Scholar] [CrossRef]
- Boräng, S.; Andersson, T.; Thelin, A.; Odeberg, J.; Lundeberg, J. Vascular gene expression in atherosclerotic plaque-prone regions analyzed by representational difference analysis. Pathobiology 2004, 71, 107–114. [Google Scholar] [CrossRef]
- Meade, J.D.; Cho, Y.J.; Fisher, J.S.; Walden, J.C.; Guo, Z.; Liang, P. Automation of fluorescent differential display with digital readout. Methods Mol. Biol. 2006, 317, 23–57. [Google Scholar] [PubMed]
- Shimkets, R.A.; Lowe, D.G.; Tai, J.T.-N.; Sehl, P.; Jin, H.; Yang, R.; Predki, P.F.; Rothberg, B.E.G.; Murtha, M.T.; Roth, M.E.; et al. Gene expression analysis by transcript profiling coupled to a gene database query. Nat. Biotechnol. 1999, 17, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.L.; Yuan, J.Y.; Zhang, J.W.; Zhang, X.H.; Wang, R.X. Differential gene expression in human hematopoietic stem cells specified toward erythroid, megakaryocytic, and granulocytic lineage. J. Leukoc. Biol. 2007, 82, 986–1002. [Google Scholar] [CrossRef]
- Carulli, J.P.; Artinger, M.; Swain, P.M.; Root, C.D.; Chee, L.; Tulig, C.; Guerin, J.; Osborne, M.; Stein, G.; Lian, J.; et al. High throughput analysis of differential gene expression. J. Cell. Biochem. 1998, 72, 286–296. [Google Scholar] [CrossRef]
- Liang, P.; Pardee, A.B. Analysing differential gene expression in cancer. Nat. Rev. Cancer 2003, 3, 869–876. [Google Scholar] [CrossRef]
- Burton, D.G.A.; Giles, P.J.; Sheerin, A.N.P.; Smith, S.K.; Lawton, J.J.; Ostler, E.L.; Rhys-Williams, W.; Kipling, D.; Faragher, R.G.A. Microarray analysis of senescent vascular smooth muscle cells: A link to atherosclerosis and vascular calcification. Exp. Gerontol. 2009, 44, 659–665. [Google Scholar] [CrossRef] [Green Version]
- Verreth, W.; De Keyzer, D.; Pelat, M.; Verhamme, P.; Ganame, J.; Bielicki, J.K.; Mertens, A.; Quarck, R.; Benhabilès, N.; Marguerie, G.; et al. Weight loss–associated induction of peroxisome proliferator–activated receptor-α and peroxisome proliferator–activated receptor-γ correlate with reduced atherosclerosis and improved cardiovascular function in obese insulin-resistant mice. Circulation 2004, 110, 3259–3269. [Google Scholar] [CrossRef] [PubMed]
- Sanger, F.; Coulson, A.R. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J. Mol. Biol. 1975, 94, 441–448. [Google Scholar] [CrossRef]
- Sim, G.K.; Kafatos, F.C.; Jones, C.W.; Koehler, M.D.; Efstratiadis, A.; Maniatis, T. Use of a cDAN library for studies on evolution and developmental expression of the chorion multigene families. Cell 1979, 18, 1303–1316. [Google Scholar] [CrossRef]
- Koch, W.J.; Ellinor, P.T.; Schwartz, A. cDNA cloning of a dihydropyridine-sensitive calcium channel from rat aorta. Evidence for the existence of alternatively spliced forms. J. Biol. Chem. 1990, 265, 17786–17791. [Google Scholar] [PubMed]
- Sohma, Y.; Suzuki, T.; Sasano, H.; Nagura, H.; Nose, M.; Yamamoto, T. Increased mRNA for CD63 antigen in atherosclerotic lesions of Watanabe heritable hyperlipidemic rabbits. Cell Struct. Funct. 1994, 19, 219–225. [Google Scholar] [CrossRef]
- Nagaraj, S.H.; Gasser, R.B.; Ranganathan, S. A hitchhiker’s guide to expressed sequence tag (est) analysis. Brief. Bioinform. 2007, 8, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Strausberg, R.L.; Feingold, E.A.; Klausner, R.D.; Collins, F.S. The mammalian gene collection. Science 1999, 286, 455–457. [Google Scholar] [CrossRef]
- Strausberg, R.L.; Feingold, E.A.; Grouse, L.H.; Derge, J.G.; Klausner, R.D.; Collins, F.S.; Wagner, L.; Shenmen, C.M.; Schuler, G.D.; Altschul, S.F.; et al. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc. Natl. Acad. Sci. USA 2002, 99, 16899–16903. [Google Scholar] [Green Version]
- de Waard, V.; van den Berg, B.M.M.; Veken, J.; Schultz-Heienbrok, R.; Pannekoek, H.; van Zonneveld, A.-J. Serial analysis of gene expression to assess the endothelial cell response to an atherogenic stimulus. Gene 1999, 226, 1–8. [Google Scholar] [CrossRef]
- Patino, W.D.; Mian, O.Y.; Kang, J.-G.; Matoba, S.; Bartlett, L.D.; Holbrook, B.; Trout, H.H.; Kozloff, L.; Hwang, P.M. Circulating transcriptome reveals markers of atherosclerosis. Proc. Natl. Acad. Sci. USA 2005, 102, 3423–3428. [Google Scholar] [CrossRef] [Green Version]
- Glenn, T.C. Field guide to next-generation DNA sequencers. Mol. Ecol. Resour. 2011, 11, 759–769. [Google Scholar] [CrossRef]
- Hashimshony, T.; Senderovich, N.; Avital, G.; Klochendler, A.; de Leeuw, Y.; Anavy, L.; Gennert, D.; Li, S.; Livak, K.J.; Rozenblatt-Rosen, O.; et al. Cel-seq2: Sensitive highly-multiplexed single-cell RNA-seq. Genome Biol. 2016, 17, 77. [Google Scholar] [CrossRef]
- Picelli, S.; Faridani, O.R.; Björklund, Å.K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-length RNA-seq from single cells using smart-seq2. Nat. Protoc. 2014, 9, 171. [Google Scholar] [CrossRef]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [Green Version]
- Ziegenhain, C.; Vieth, B.; Parekh, S.; Reinius, B.; Guillaumet-Adkins, A.; Smets, M.; Leonhardt, H.; Heyn, H.; Hellmann, I.; Enard, W. Comparative analysis of single-cell RNA sequencing methods. Mol. Cell 2017, 65, 631–643.e4. [Google Scholar] [CrossRef] [PubMed]
- Cochain, C.; Vafadarnejad, E.; Arampatzi, P.; Pelisek, J.; Winkels, H.; Ley, K.; Wolf, D.; Saliba, A.E.; Zernecke, A. Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ. Res. 2018, 122, 1661–1674. [Google Scholar] [CrossRef] [PubMed]
- Brodin, J.; Mild, M.; Hedskog, C.; Sherwood, E.; Leitner, T.; Andersson, B.; Albert, J. PCR-induced transitions are the major source of error in cleaned ultra-deep pyrosequencing data. PLoS ONE 2013, 8, e70388. [Google Scholar] [CrossRef] [PubMed]
- Poptsova, M.S.; Il’icheva, I.A.; Nechipurenko, D.Y.; Panchenko, L.A.; Khodikov, M.V.; Oparina, N.Y.; Polozov, R.V.; Nechipurenko, Y.D.; Grokhovsky, S.L. Non-random DNA fragmentation in next-generation sequencing. Sci. Rep. 2014, 4, 4532. [Google Scholar] [CrossRef] [PubMed]
- Quail, M.A.; Smith, M.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.P.; Gu, Y. A tale of three next generation sequencing platforms: Comparison of ion torrent, pacific biosciences and Illumina miseq sequencers. BMC Genom. 2012, 13, 341. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Jhaveri, D.J.; Marshall, V.M.; Bauer, D.C.; Edson, J.; Narayanan, R.K.; Robinson, G.J.; Lundberg, A.E.; Bartlett, P.F.; Wray, N.R.; et al. A comparative study of techniques for differential expression analysis on RNA-seq data. PLoS ONE 2014, 9, e103207. [Google Scholar] [CrossRef]
- Rapaport, F.; Khanin, R.; Liang, Y.; Pirun, M.; Krek, A.; Zumbo, P.; Mason, C.E.; Socci, N.D.; Betel, D. Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data. Genome Biol. 2013, 14, 3158. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhou, J.; White, K.P. RNA-seq differential expression studies: More sequence or more replication? Bioinformatics 2014, 30, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Schurch, N.J.; Schofield, P.; Gierliński, M.; Cole, C.; Sherstnev, A.; Singh, V.; Wrobel, N.; Gharbi, K.; Simpson, G.G.; Owen-Hughes, T.; et al. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? RNA 2016, 22, 839–851. [Google Scholar] [CrossRef] [Green Version]
- Chhangawala, S.; Rudy, G.; Mason, C.E.; Rosenfeld, J.A. The impact of read length on quantification of differentially expressed genes and splice junction detection. Genome Biol. 2015, 16, 131. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, Y.; Gamini, R.; Zhang, B.; von Schack, D. Evaluation of two main RNA-seq approaches for gene quantification in clinical rna sequencing: Polya+ selection versus rrna depletion. Sci. Rep. 2018, 8, 4781. [Google Scholar] [CrossRef] [PubMed]
- Raplee, I.D.; Evsikov, A.V.; Marin de Evsikova, C. Aligning the aligners: Comparison of rna sequencing data alignment and gene expression quantification tools for clinical breast cancer research. J. Pers. Med. 2019, 9, 18. [Google Scholar] [CrossRef]
- Eikrem, O.; Beisland, C.; Hjelle, K.; Flatberg, A.; Scherer, A.; Landolt, L.; Skogstrand, T.; Leh, S.; Beisvag, V.; Marti, H.-P. Transcriptome sequencing (rnaseq) enables utilization of formalin-fixed, paraffin-embedded biopsies with clear cell renal cell carcinoma for exploration of disease biology and biomarker development. PLoS ONE 2016, 11, e0149743. [Google Scholar] [CrossRef] [PubMed]
- Esteve-Codina, A.; Arpi, O.; Martinez-García, M.; Pineda, E.; Mallo, M.; Gut, M.; Carrato, C.; Rovira, A.; Lopez, R.; Tortosa, A.; et al. A comparison of RNA-seq results from paired formalin-fixed paraffin-embedded and fresh-frozen glioblastoma tissue samples. PLoS ONE 2017, 12, e0170632. [Google Scholar] [CrossRef] [PubMed]
- Brunner, A.L.; Li, J.; Guo, X.; Sweeney, R.T.; Varma, S.; Zhu, S.X.; Li, R.; Tibshirani, R.; West, R.B. A shared transcriptional program in early breast neoplasias despite genetic and clinical distinctions. Genome Biol. 2014, 15, R71. [Google Scholar] [CrossRef] [PubMed]
- Nookaew, I.; Papini, M.; Pornputtapong, N.; Scalcinati, G.; Fagerberg, L.; Uhlén, M.; Nielsen, J. A comprehensive comparison of RNA-seq-based transcriptome analysis from reads to differential gene expression and cross-comparison with microarrays: A case study in saccharomyces cerevisiae. Nucleic Acids Res. 2012, 40, 10084–10097. [Google Scholar] [CrossRef]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef]
- Mosher, J.J.; Bowman, B.; Bernberg, E.L.; Shevchenko, O.; Kan, J.; Korlach, J.; Kaplan, L.A. Improved performance of the pacbio smrt technology for 16s rdna sequencing. J. Microbiol. Methods 2014, 104, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.; Au, K.F. Pacbio sequencing and its applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Bayega, A.; Wang, Y.C.; Oikonomopoulos, S.; Djambazian, H.; Fahiminiya, S.; Ragoussis, J. Transcript profiling using long-read sequencing technologies. In Gene Expression Analysis: Methods and Protocols; Raghavachari, N., Garcia-Reyero, N., Eds.; Springer: New York, NY, USA, 2018; pp. 121–147. [Google Scholar]
- Gordon, D.; Huddleston, J.; Chaisson, M.J.P.; Hill, C.M.; Kronenberg, Z.N.; Munson, K.M.; Malig, M.; Raja, A.; Fiddes, I.; Hillier, L.W.; et al. Long-read sequence assembly of the gorilla genome. Science 2016, 352, aae0344. [Google Scholar] [CrossRef]
- Quackenbush, J. Microarray data normalization and transformation. Nat. Genet. 2002, 32, 496–501. [Google Scholar] [CrossRef]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. Affy—Analysis of Affymetrix genechip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Kibbe, W.A.; Lin, S.M. Lumi: A pipeline for processing illumina microarray. Bioinformatics 2008, 24, 1547–1548. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Bioconductor. Available online: https://www.bioconductor.org/ (accessed on 27 April 2019).
- Andrews, S. Fastqc a Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 27 April 2019).
- Dai, M.; Thompson, R.C.; Maher, C.; Contreras-Galindo, R.; Kaplan, M.H.; Markovitz, D.M.; Omenn, G.; Meng, F. NGSQC: Cross-platform quality analysis pipeline for deep sequencing data. BMC Genom. 2010, 11, S7. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A.; Hannon, G.J. Fastx-Toolkit. Available online: http://hannonlab.cshl.edu/fastx_toolkit/ (accessed on 27 April 2019).
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. Star: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. Featurecounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Eilbeck, K.; Lewis, S.E.; Mungall, C.J.; Yandell, M.; Stein, L.; Durbin, R.; Ashburner, M. The sequence ontology: A tool for the unification of genome annotations. Genome Biol. 2005, 6, R44. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Carbon, S.; Ireland, A.; Mungall, C.J.; Shu, S.; Marshall, B.; Lewis, S.; AmiGO Hub and Web Presence Working Group. Amigo: Online access to ontology and annotation data. Bioinformatics 2009, 25, 288–289. [Google Scholar] [CrossRef]
- Lovering, R.C.; Roncaglia, P.; Howe, D.G.; Laulederkind, S.J.F.; Khodiyar, V.K.; Berardini, T.Z.; Tweedie, S.; Foulger, R.E.; Osumi-Sutherland, D.; Campbell, N.H.; et al. Improving interpretation of cardiac phenotypes and enhancing discovery with expanded knowledge in the gene ontology. Circ. Genom. Precis. Med. 2018, 11, e001813. [Google Scholar] [CrossRef]
- Smith, C.L.; Goldsmith, C.-A.W.; Eppig, J.T. The mammalian phenotype ontology as a tool for annotating, analyzing and comparing phenotypic information. Genome Biol. 2004, 6, R7. [Google Scholar] [CrossRef]
- Arze, C.; Feng, G.; Mazaitis, M.; Nadendla, S.; Felix, V.; Chang, Y.-W.W.; Schriml, L.M.; Kibbe, W.A. Disease ontology: A backbone for disease semantic integration. Nucleic Acids Res. 2011, 40, D940–D946. [Google Scholar]
- Bello, S.M.; Shimoyama, M.; Mitraka, E.; Laulederkind, S.J.F.; Smith, C.L.; Eppig, J.T.; Schriml, L.M. Disease ontology: Improving and unifying disease annotations across species. Dis. Models Mech. 2018, 11, dmm032839. [Google Scholar] [CrossRef]
- Bult, C.J.; Drabkin, H.J.; Evsikov, A.; Natale, D.; Arighi, C.; Roberts, N.; Ruttenberg, A.; D’Eustachio, P.; Smith, B.; Blake, J.A.; et al. The representation of protein complexes in the protein ontology (pro). BMC Bioinform. 2011, 12, 371. [Google Scholar] [CrossRef]
- Natale, D.A.; Arighi, C.N.; Barker, W.C.; Blake, J.A.; Bult, C.J.; Caudy, M.; Drabkin, H.J.; D’Eustachio, P.; Evsikov, A.V.; Huang, H.; et al. The protein ontology: A structured representation of protein forms and complexes. Nucleic Acids Res. 2011, 39, D539–D545. [Google Scholar] [CrossRef]
- Smith, B.; Ashburner, M.; Rosse, C.; Bard, J.; Bug, W.; Ceusters, W.; Goldberg, L.J.; Eilbeck, K.; Ireland, A.; Mungall, C.J.; et al. The obo foundry: Coordinated evolution of ontologies to support biomedical data integration. Nat. Biotechnol. 2007, 25, 1251–1255. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Caspi, R.; Billington, R.; Ferrer, L.; Foerster, H.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Mueller, L.A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2016, 44, D471–D480. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. Kegg: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Richardson, J.E.; Bult, C.J. Visual annotation display (VLAD): A tool for finding functional themes in lists of genes. Mamm. Genome 2015, 26, 567–573. [Google Scholar] [CrossRef]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. Gorilla: A tool for discovery and visualization of enriched go terms in ranked gene lists. BMC Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef]
- Jiao, X.; Sherman, B.T.; Huang, D.W.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. David-ws: A stateful web service to facilitate gene/protein list analysis. Bioinformatics 2012, 28, 1805–1806. [Google Scholar] [CrossRef]
- Maere, S.; Heymans, K.; Kuiper, M. Bingo: A cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef]
- Evsikov, A.V.; Marín de Evsikova, C. Transcriptomics as precision medicine to classify in vivo models of dietary-induced atherosclerosis at cellular and molecular levels. J. Pers. Med. 2018. [Google Scholar] [CrossRef]
- Krämer, A.; Green, J.; Pollard, J.J.; Tugendreich, S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Nikitin, A.; Egorov, S.; Daraselia, N.; Mazo, I. Pathway studio—The analysis and navigation of molecular networks. Bioinformatics 2003, 19, 2155–2157. [Google Scholar] [CrossRef]
- Romero, P.; Wagg, J.; Green, M.L.; Kaiser, D.; Krummenacker, M.; Karp, P.D. Computational prediction of human metabolic pathways from the complete human genome. Genome Biol. 2004, 6, R2. [Google Scholar] [CrossRef]
- Evsikov, A.V.; Dolan, M.E.; Genrich, M.P.; Patek, E.; Bult, C.J. Mousecyc: A curated biochemical pathways database for the laboratory mouse. Genome Biol. 2009, 10, R84. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Lewin, H.A. Reconstruction of metabolic pathways for the cattle genome. BMC Syst. Biol. 2009, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Cieślik, M.; Chinnaiyan, A.M. Cancer transcriptome profiling at the juncture of clinical translation. Nat. Rev. Genet. 2017, 19, 93. [Google Scholar] [CrossRef]
- Gatsiou, A.; Stellos, K. Dawn of epitranscriptomic medicine. Circ. Genom. Precis. Med. 2018, 11, e001927. [Google Scholar] [CrossRef]
- Barrett, T.J.; Lee, A.H.; Smilowitz, N.R.; Hausvater, A.; Fishman, G.I.; Hochman, J.S.; Reynolds, H.R.; Berger, J.S. Whole-blood transcriptome profiling identifies women with myocardial infarction with nonobstructive coronary artery disease. Circ. Genom. Precis. Med. 2018, 11, e002387. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K.; Bernstein, D.; Cole, F.S.; Khokha, M.K.; Lee, F.S.; Lin, S.; McDonald, T.V.; Moskowitz, I.P.; Quertermous, T.; Sankaran, V.G.; et al. Functional assays to screen and dissect genomic hits. Circ. Genom. Precis. Med. 2018, 11, e002178. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Resource | Description | URL | Ref |
|---|---|---|---|
| Databases: | |||
| Gene Ontology | Central repository of terms describing gene functions across multiple biological systems | http://geneontology.org/ | [77] |
| Mammalian Phenotype Ontology | Biomedical curators’ and community database of ontological terms for annotating phenotypic data | http://www.informatics.jax.org/vocab/mp_ontology/ | [80] |
| Human Disease Ontology | Ontology for human disease cross-mapped to MeSH, ICD, NCI’s thesaurus, SNOMED and OMIM. | http://disease-ontology.org/ | [81] |
| Protein Ontology | Ontology of protein-related entities, their explicit definitions, and relationships between them. | https://proconsortium.org/pro/pro.shtml | [84] |
| Open Biological Ontologies | Collaborative effort to specify and implement best principles and practices in ontology development. Contains links to all Ontologies. | http://obofoundry.org/ | [85] |
| MSigDB | A collection of annotated gene sets, such as canonical pathways gene sets, for use with GSEA. | http://software.broadinstitute.org/gsea/msigdb/index.jsp | [86] |
| MetaCyc | A curated database of experimentally elucidated metabolic pathways for many organisms. | https://metacyc.org/ | [87] |
| KEGG | A collection of maps representing metabolism, pathways, and associated genes. | https://www.genome.jp/kegg/ | [88] |
| Reactome | A free, open-source, curated and peer-reviewed pathway database. | https://reactome.org/ | [89] |
| Tools: | |||
| VLAD | Tool for identification of statistically significant over- or under-represented ontology terms in lists of genes. GO gene – function annotations for human and mouse, and MP gene – phenotype annotations for mouse are pre-loaded. Allows uploading user-specified ontologies and gene – ontology mappings. Updated weekly. | http://proto.informatics.jax.org/prototypes/vlad/ | [90] |
| AmiGO | Allows users to query, browse and visualize ontologies and gene annotation data for many species. Updated weekly. | http://amigo.geneontology.org/amigo | [78] |
| GOrilla | A tool to identify and visualize enriched GO terms in gene lists. Can either search for GO terms at the top of a ranked gene list, or compare a target gene list to a background gene list. | http://cbl-gorilla.cs.technion.ac.il/ | [91] |
| DAVID | A set of tools to identify overrepresented features in large lists of genes. | https://david.ncifcrf.gov/ | [92] |
| BinGO | Cytoscape tool to visualize statistically overrepresented GO terms in a list of genes. | http://apps.cytoscape.org/apps/bingo1 | [93] |
| GSEA | A tool to determine if a gene set has significant differences between two biological states. | http://software.broadinstitute.org/gsea/downloads.jsp1 | [86] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marín de Evsikova, C.; Raplee, I.D.; Lockhart, J.; Jaimes, G.; Evsikov, A.V. The Transcriptomic Toolbox: Resources for Interpreting Large Gene Expression Data within a Precision Medicine Context for Metabolic Disease Atherosclerosis. J. Pers. Med. 2019, 9, 21. https://doi.org/10.3390/jpm9020021
Marín de Evsikova C, Raplee ID, Lockhart J, Jaimes G, Evsikov AV. The Transcriptomic Toolbox: Resources for Interpreting Large Gene Expression Data within a Precision Medicine Context for Metabolic Disease Atherosclerosis. Journal of Personalized Medicine. 2019; 9(2):21. https://doi.org/10.3390/jpm9020021
Chicago/Turabian StyleMarín de Evsikova, Caralina, Isaac D. Raplee, John Lockhart, Gilberto Jaimes, and Alexei V. Evsikov. 2019. "The Transcriptomic Toolbox: Resources for Interpreting Large Gene Expression Data within a Precision Medicine Context for Metabolic Disease Atherosclerosis" Journal of Personalized Medicine 9, no. 2: 21. https://doi.org/10.3390/jpm9020021