VARIFI—Web-Based Automatic Variant Identification, Filtering and Annotation of Amplicon Sequencing Data

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation and Amplicon Sequencing
2.2. Bioinformatic Analysis
2.2.1. Read Alignment
2.2.2. Alignment Post-Processing
2.2.3. Variant Identification
2.2.4. Variant Filtration
2.2.5. Merging Variants
2.2.6. Variant Annotation
2.2.7. Visualization
2.2.8. Amplicon Coverage and Plotting
2.3. Variant Validation—Sanger Sequencing
2.4. VAIRFI Performance Calculations and Comparison with Ion Reporter
2.5. Ethics Committee Approval
3. Results
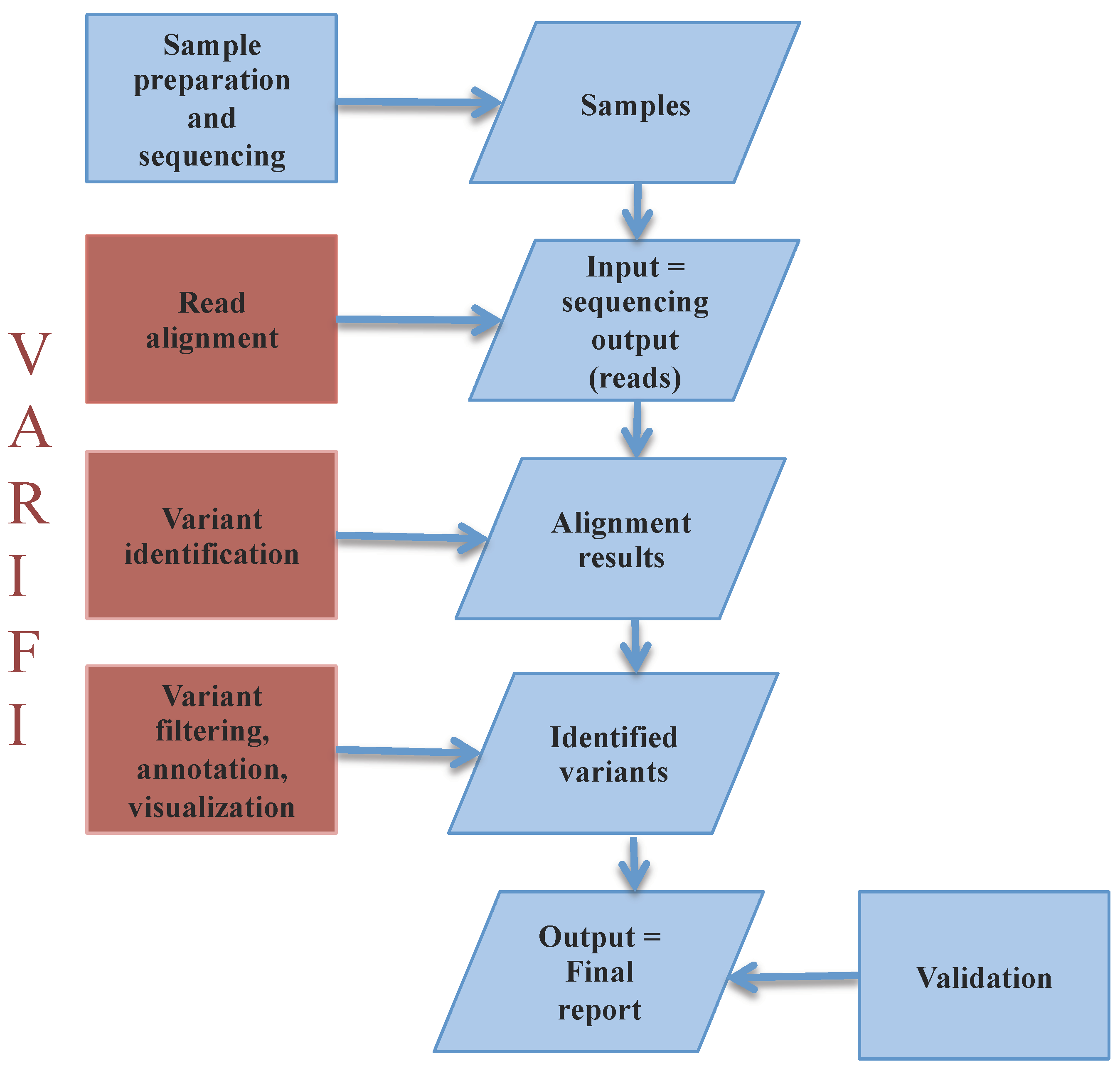
3.1. VARIFI Pipeline
3.1.1. Input Files
3.1.2. Processing Files
3.1.3. Variant Filtering
3.1.4. Confidence Score
3.1.5. Variant Annotation
3.1.6. Output Files
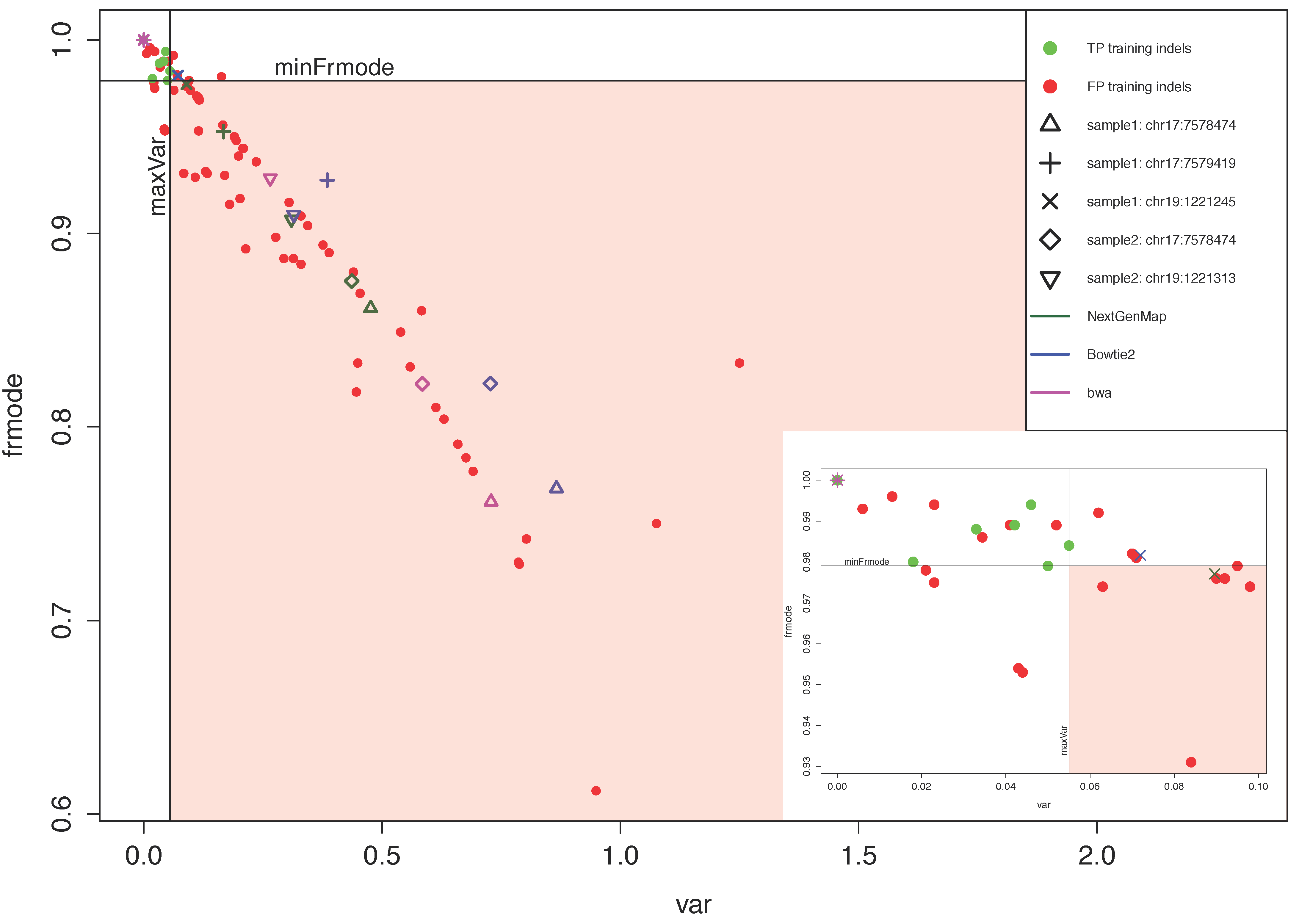
3.2. VARIFI Validation and Evaluation
3.2.1. Input File Examples
3.2.2. VARIFI Performance Evaluation
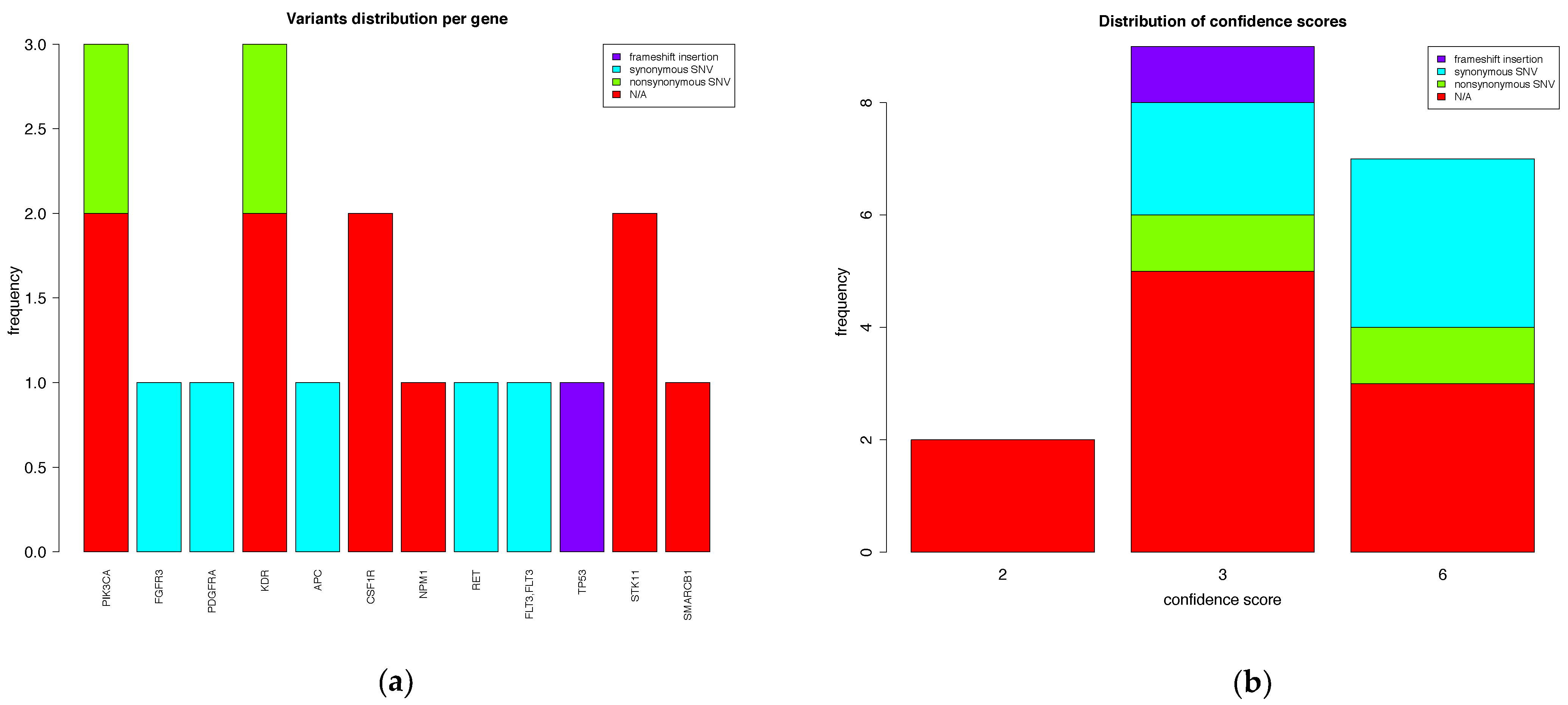
3.2.3. Output File Examples
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Meldrum, C.; Doyle, M.A.; Tothill, R.W. Next-generation sequencing for cancer diagnostics: A practical perspective. Clin. Biochem. Rev. Aust. Assoc. Clin. Biochem. 2011, 32, 177–195. [Google Scholar]
- Rehm, H.L. Disease-targeted sequencing: A cornerstone in the clinic. Nat. Rev. Genet. 2013, 14, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Mendez, P.; Dang, J.; Kim, J.W.; Lee, S.; Yoon, J.H.; Kim, T.; Sailey, C.J.; Jablons, D.M.; Kim, I.J. Comprehensive evaluation and validation of targeted next-generation sequencing performance in two clinical laboratories. Int. J. Oncol. 2016, 49, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, F.C.; Voss, J.S.; Kipp, B.R.; Kerr, S.E.; Van Arnam, J.S.; Mills, J.R.; Marcou, C.A.; Schneider, A.R.; Tu, Z.J.; Henry, M.R.; et al. Assessment of pancreatic neuroendocrine tumor cytologic genotype diversity to guide personalized medicine using a custom gastroenteropancreatic next-generation sequencing panel. Oncotarget 2017, 8, 93464–93475. [Google Scholar] [CrossRef] [PubMed]
- Ewing, A.D.; Houlahan, K.E.; Hu, Y.; Ellrott, K.; Caloian, C.; Yamaguchi, T.N.; Bare, J.C.; P’ng, C.; Waggott, D.; Sabelnykova, V.Y.; et al. Combining tumor genome simulation with crowdsourcing to benchmark somatic single-nucleotide-variant detection. Nat. Methods 2015, 12, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Alioto, T.S.; Buchhalter, I.; Derdak, S.; Hutter, B.; Eldridge, M.D.; Hovig, E.; Heisler, L.E.; Beck, T.A.; Simpson, J.T.; Tonon, L.; et al. A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing. Nat. Commun. 2015, 6, 10001. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Sedlazeck, F.J.; Rescheneder, P.; von Haeseler, A. Nextgenmap: Fast and accurate read mapping in highly polymorphic genomes. Bioinformatics 2013, 29, 2790–2791. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing Subgroup. The sequence alignment/map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. Annovar: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Genomes Project, C.; Abecasis, G.R.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; Handsaker, R.E.; Kang, H.M.; Marth, G.T.; McVean, G.A. An integrated map of genetic variation from 1,092 human genomes. Nature 2012, 491, 56–65. [Google Scholar]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Rodelsperger, C.; Schuelke, M.; Seelow, D. Mutationtaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [PubMed]
- Davydov, E.V.; Goode, D.L.; Sirota, M.; Cooper, G.M.; Sidow, A.; Batzoglou, S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the sift algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Beare, D.; Gunasekaran, P.; Leung, K.; Bindal, N.; Boutselakis, H.; Ding, M.; Bamford, S.; Cole, C.; Ward, S.; et al. Cosmic: Exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015, 43, D805–D811. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012, 2012, 251364. [Google Scholar] [CrossRef] [PubMed]
- Bragg, L.M.; Stone, G.; Butler, M.K.; Hugenholtz, P.; Tyson, G.W. Shining a light on dark sequencing: Characterising errors in ion torrent pgm data. PLoS Comput. Biol. 2013, 9, e1003031. [Google Scholar] [CrossRef] [PubMed]
- Yeo, Z.X.; Chan, M.; Yap, Y.S.; Ang, P.; Rozen, S.; Lee, A.S. Improving indel detection specificity of the ion Torrent PGM benchtop sequencer. PLoS ONE 2012, 7, e45798. [Google Scholar] [CrossRef] [PubMed]
- Yeo, Z.X.; Wong, J.C.; Rozen, S.G.; Lee, A.S. Evaluation and optimisation of indel detection workflows for ion torrent sequencing of the BRCA1 and BRCA2 genes. BMC Genom. 2014, 15, 516. [Google Scholar] [CrossRef] [PubMed]
- Zook, J.M.; Chapman, B.; Wang, J.; Mittelman, D.; Hofmann, O.; Hide, W.; Salit, M. Integrating human sequence data sets provides a resource of benchmark snp and indel genotype calls. Nat. Biotechnol. 2014, 32, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Mu, W.; Lu, H.M.; Chen, J.; Li, S.; Elliott, A.M. Sanger confirmation is required to achieve optimal sensitivity and specificity in next-generation sequencing panel testing. J. Mol. Diagn. JMD 2016, 18, 923–932. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krunic, M.; Venhuizen, P.; Müllauer, L.; Kaserer, B.; von Haeseler, A. VARIFI—Web-Based Automatic Variant Identification, Filtering and Annotation of Amplicon Sequencing Data. J. Pers. Med. 2019, 9, 10. https://doi.org/10.3390/jpm9010010
Krunic M, Venhuizen P, Müllauer L, Kaserer B, von Haeseler A. VARIFI—Web-Based Automatic Variant Identification, Filtering and Annotation of Amplicon Sequencing Data. Journal of Personalized Medicine. 2019; 9(1):10. https://doi.org/10.3390/jpm9010010
Chicago/Turabian StyleKrunic, Milica, Peter Venhuizen, Leonhard Müllauer, Bettina Kaserer, and Arndt von Haeseler. 2019. "VARIFI—Web-Based Automatic Variant Identification, Filtering and Annotation of Amplicon Sequencing Data" Journal of Personalized Medicine 9, no. 1: 10. https://doi.org/10.3390/jpm9010010