
Pharmacogenetics in the Treatment of Huntington’s Disease: Review and Future Perspectives
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
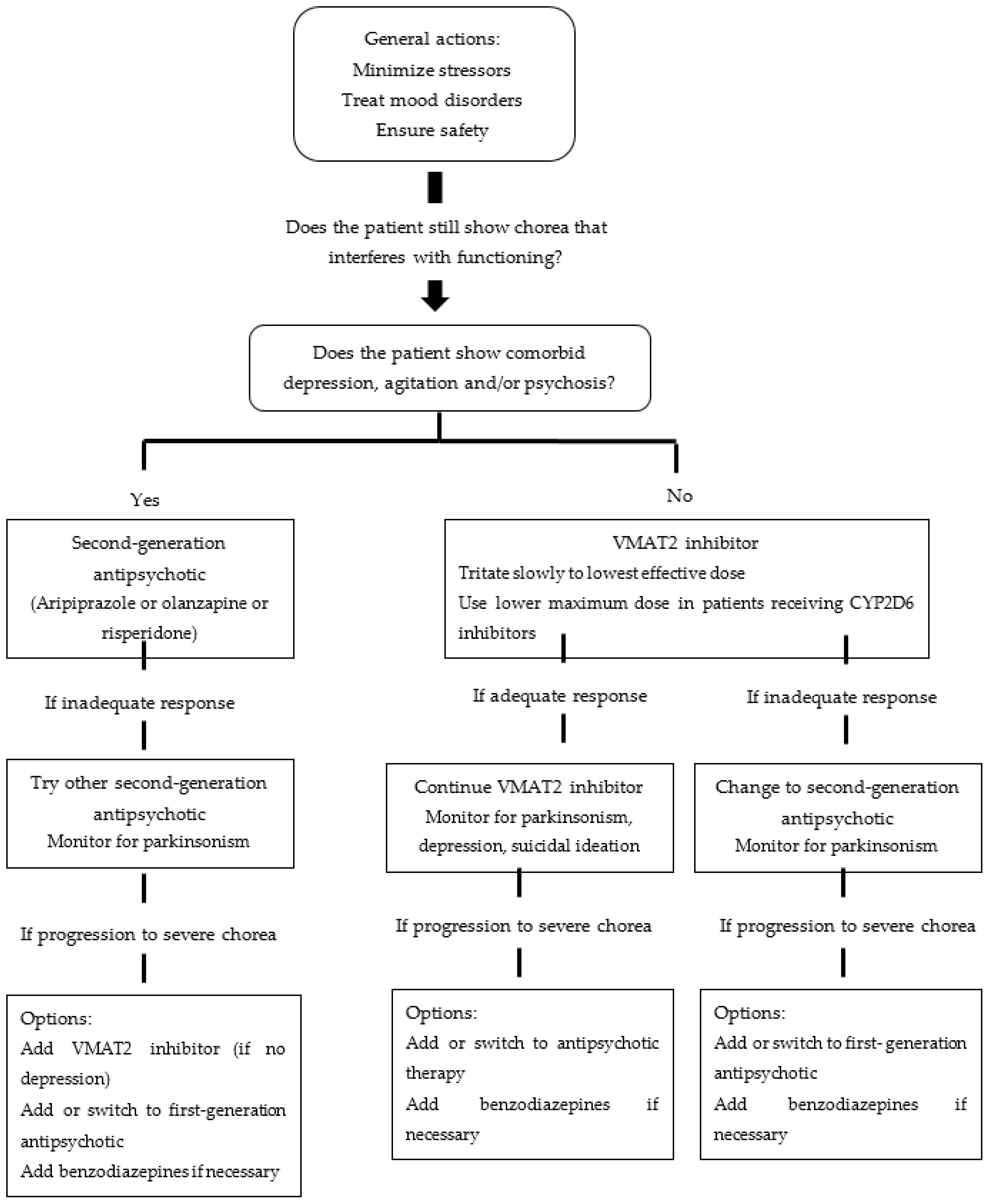
3. Pharmacogenetics of the Drugs Used in the Management of Chorea in Huntington’s Disease
3.1. Monoamine Transporter Type 2 (VMAT2) Inhibitors (Tetrabenazine and Deutetrabenazine)
3.2. Second-Generation Antipsychotic Drugs (Aripiprazole, Olanzapine, and Risperidone)
3.3. First-Generation Antipsychotic Drugs (Haloperidol)
3.4. Other Drugs Used in the Management of Chorea
4. Pharmacogenetics of the Drugs Used in the Management of Depression, Irritability, Apathy, Anxiety and Psychosis in Huntington’s Disease
5. Other Biomarkers in Study
6. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Post, A.E.M.; Klockgether, T.; Landwehrmeyer, G.B.; Pandolfo, M.; Arnesen, A.; Reinhard, C.; Graessner, H. Research Priorities for Rare Neurological Diseases: A Representative View of Patient Representatives and Healthcare Professionals from the European Reference Network for Rare Neurological Diseases. Orphanet J. Rare Dis. 2021, 16, 135. [Google Scholar] [CrossRef]
- Pringsheim, T.; Wiltshire, K.; Day, L.; Dykeman, J.; Steeves, T.; Jette, N. The Incidence and Prevalence of Huntington’s Disease: A Systematic Review and Meta-Analysis. Mov. Disord. Off. J. Mov. Disord. Soc. 2012, 27, 1083–1091. [Google Scholar] [CrossRef]
- Tortelli, R.; Seripa, D.; Panza, F.; Solfrizzi, V.; Logroscino, G. Pharmacogenetics in Neurodegenerative Diseases: Implications for Clinical Trials. In Frontiers of Neurology and Neuroscience; Beghi, E., Logroscino, G., Eds.; S. Karger AG: Berlin, Germany, 2016; Volume 39, pp. 124–135. ISBN 978-3-318-05864-2. [Google Scholar]
- Armstrong, M.J.; Miyasaki, J.M. American Academy of Neurology Evidence-Based Guideline: Pharmacologic Treatment of Chorea in Huntington Disease: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology 2012, 79, 597–603. [Google Scholar] [CrossRef] [Green Version]
- Spear, B.B.; Heath-Chiozzi, M.; Huff, J. Clinical Application of Pharmacogenetics. Trends Mol. Med. 2001, 7, 201–204. [Google Scholar] [CrossRef]
- Bouvy, J.C.; De Bruin, M.L.; Koopmanschap, M.A. Epidemiology of Adverse Drug Reactions in Europe: A Review of Recent Observational Studies. Drug Saf. 2015, 38, 437–453. [Google Scholar] [CrossRef] [Green Version]
- Mejía, G.; Saiz-Rodríguez, M.; Gómez de Olea, B.; Ochoa, D.; Abad-Santos, F. Urgent Hospital Admissions Caused by Adverse Drug Reactions and Medication Errors—A Population-Based Study in Spain. Front. Pharmacol. 2020, 11, 734. [Google Scholar] [CrossRef]
- Sultana, J.; Cutroneo, P.; Trifirò, G. Clinical and Economic Burden of Adverse Drug Reactions. J. Pharmacol. Pharmacother. 2013, 4, 73. [Google Scholar] [CrossRef] [Green Version]
- Siafis, S.; Tzachanis, D.; Samara, M.; Papazisis, G. Antipsychotic Drugs: From Receptor-Binding Profiles to Metabolic Side Effects. Curr. Neuropharmacol. 2018, 16, 1210–1223. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. PRISMA Group Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. J. Clin. Epidemiol. 2009, 62, 1006–1012. [Google Scholar] [CrossRef]
- Whirl-Carrillo, M.; McDonagh, E.M.; Hebert, J.M.; Gong, L.; Sangkuhl, K.; Thorn, C.F.; Altman, R.B.; Klein, T.E. Pharmacogenomics Knowledge for Personalized Medicine. Clin. Pharmacol. Ther. 2012, 92, 414–417. [Google Scholar] [CrossRef]
- Bachoud-Lévi, A.-C.; Ferreira, J.; Massart, R.; Youssov, K.; Rosser, A.; Busse, M.; Craufurd, D.; Reilmann, R.; De Michele, G.; Rae, D.; et al. International Guidelines for the Treatment of Huntington’s Disease. Front. Neurol. 2019, 10, 710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suchowersky, O. Huntington Disease: Management. Available online: https://www.uptodate.com/contents/huntington-disease-management (accessed on 11 January 2023).
- Schneider, F.; Stamler, D.; Bradbury, M.; Loupe, P.S.; Hellriegel, E.; Cox, D.S.; Savola, J.-M.; Gordon, M.F.; Rabinovich-Guilatt, L. Pharmacokinetics of Deutetrabenazine and Tetrabenazine: Dose Proportionality and Food Effect. Clin. Pharmacol. Drug Dev. 2021, 10, 647–659. [Google Scholar] [CrossRef]
- Huntington Study Group. Tetrabenazine as Antichorea Therapy in Huntington Disease: A Randomized Controlled Trial. Neurology 2006, 66, 366–372. [Google Scholar] [CrossRef]
- Frank, S.; Testa, C.; Edmondson, M.C.; Goldstein, J.; Kayson, E.; Leavitt, B.R.; Oakes, D.; O’Neill, C.; Vaughan, C.; Whaley, J.; et al. The Safety of Deutetrabenazine for Chorea in Huntington Disease: An Open-Label Extension Study. CNS Drugs 2022, 36, 1207–1216. [Google Scholar] [CrossRef]
- Jankovic, J.; Clarence-Smith, K. Tetrabenazine for the Treatment of Chorea and Other Hyperkinetic Movement Disorders. Expert Rev. Neurother. 2011, 11, 1509–1523. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M.; Sim, S.C.; Gomez, A.; Rodriguez-Antona, C. Influence of Cytochrome P450 Polymorphisms on Drug Therapies: Pharmacogenetic, Pharmacoepigenetic and Clinical Aspects. Pharmacol. Ther. 2007, 116, 496–526. [Google Scholar] [CrossRef]
- The Pharmacogene Variation (PharmVar) Consortium. CYP2D6 Allele Nomenclature. Available online: https://www.pharmvar.org/gene/CYP2D6 (accessed on 20 February 2023).
- Caudle, K.E.; Dunnenberger, H.M.; Freimuth, R.R.; Peterson, J.F.; Burlison, J.D.; Whirl-Carrillo, M.; Scott, S.A.; Rehm, H.L.; Williams, M.S.; Klein, T.E.; et al. Standardizing Terms for Clinical Pharmacogenetic Test Results: Consensus Terms from the Clinical Pharmacogenetics Implementation Consortium (CPIC). Genet. Med. Off. J. Am. Coll. Med. Genet. 2017, 19, 215–223. [Google Scholar] [CrossRef]
- Clinical Pharmacology. Department of Medicine. Indiana University. Available online: https://drug-interactions.medicine.iu.edu/maintable.aspx (accessed on 10 January 2023).
- EMA Scientific Conclusions and Grounds for Variation, Amendments to the Product Information of Tetrabenazine. Available online: https://www.ema.europa.eu/en/documents/psusa/tetrabenazinecmdh-scientific-conclusions-grounds-variation-amendments-product-information-timetable/00002911/201410_en.pdf (accessed on 24 January 2023).
- Mehanna, R.; Hunter, C.; Davidson, A.; Jimenez-Shahed, J.; Jankovic, J. Analysis of CYP2D6 Genotype and Response to Tetrabenazine. Mov. Disord. Off. J. Mov. Disord. Soc. 2013, 28, 210–215. [Google Scholar] [CrossRef]
- Ciammola, A.; Sassone, J.; Colciago, C.; Mencacci, N.E.; Poletti, B.; Ciarmiello, A.; Squitieri, F.; Silani, V. Aripiprazole in the Treatment of Huntington’s Disease: A Case Series. Neuropsychiatr. Dis. Treat. 2009, 5, 1–4. [Google Scholar]
- Brusa, L.; Orlacchio, A.; Moschella, V.; Iani, C.; Bernardi, G.; Mercuri, N.B. Treatment of the Symptoms of Huntington’s Disease: Preliminary Results Comparing Aripiprazole and Tetrabenazine. Mov. Disord. Off. J. Mov. Disord. Soc. 2009, 24, 126–129. [Google Scholar] [CrossRef]
- Bonelli, R.M.; Mahnert, F.A.; Niederwieser, G. Olanzapine for Huntington’s Disease: An Open Label Study. Clin. Neuropharmacol. 2002, 25, 263–265. [Google Scholar] [CrossRef]
- Erdemoglu, A.K.; Boratav, C. Risperidone in Chorea and Psychosis of Huntington’s Disease. Eur. J. Neurol. 2002, 9, 182–183. [Google Scholar] [CrossRef]
- Gupta, S.; Masand, P. Aripiprazole: Review of Its Pharmacology and Therapeutic Use in Psychiatric Disorders. Ann. Clin. Psychiatry Off. J. Am. Acad. Clin. Psychiatr. 2004, 16, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Bourin, M.; Baker, G.B. Metabolism of Risperidone to 9-Hydroxyrisperidone by Human Cytochromes P450 2D6 and 3A4. Naunyn. Schmiedebergs Arch. Pharmacol. 1999, 359, 147–151. [Google Scholar] [CrossRef]
- Kiss, Á.; Menus, Á.; Tóth, K.; Déri, M.; Sirok, D.; Gabri, E.; Belic, A.; Csukly, G.; Bitter, I.; Monostory, K. Phenoconversion of CYP2D6 by Inhibitors Modifies Aripiprazole Exposure. Eur. Arch. Psychiatry Clin. Neurosci. 2020, 270, 71–82. [Google Scholar] [CrossRef] [Green Version]
- Hendset, M.; Hermann, M.; Lunde, H.; Refsum, H.; Molden, E. Impact of the CYP2D6 Genotype on Steady-State Serum Concentrations of Aripiprazole and Dehydroaripiprazole. Eur. J. Clin. Pharmacol. 2007, 63, 1147–1151. [Google Scholar] [CrossRef]
- van der Weide, K.; van der Weide, J. The Influence of the CYP3A4*22 Polymorphism and CYP2D6 Polymorphisms on Serum Concentrations of Aripiprazole, Haloperidol, Pimozide, and Risperidone in Psychiatric Patients. J. Clin. Psychopharmacol. 2015, 35, 228–236. [Google Scholar] [CrossRef]
- Lisbeth, P.; Vincent, H.; Kristof, M.; Bernard, S.; Manuel, M.; Hugo, N. Genotype and Co-Medication Dependent CYP2D6 Metabolic Activity: Effects on Serum Concentrations of Aripiprazole, Haloperidol, Risperidone, Paliperidone and Zuclopenthixol. Eur. J. Clin. Pharmacol. 2016, 72, 175–184. [Google Scholar] [CrossRef]
- U.S. Food & Drug Administration (FDA). Drugs FDA: FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&varapplno=021436 (accessed on 11 January 2023).
- European Medicines Agency (EMA). Abilify Maintena. Available online: https://www.ema.europa.eu/en/medicines/human/epar/abilify-maintena (accessed on 11 January 2023).
- Swen, J.J.; Nijenhuis, M.; de Boer, A.; Grandia, L.; Maitland-van der Zee, A.H.; Mulder, H.; Rongen, G.A.P.J.M.; van Schaik, R.H.N.; Schalekamp, T.; Touw, D.J.; et al. Pharmacogenetics: From Bench to Byte—An Update of Guidelines. Clin. Pharmacol. Ther. 2011, 89, 662–673. [Google Scholar] [CrossRef]
- Shen, Y.-C.; Chen, S.-F.; Chen, C.-H.; Lin, C.C.H.; Chen, S.-J.; Chen, Y.-J.; Luu, S.-U. Effects of DRD2/ANKK1 Gene Variations and Clinical Factors on Aripiprazole Efficacy in Schizophrenic Patients. J. Psychiatr. Res. 2009, 43, 600–606. [Google Scholar] [CrossRef]
- Kwon, J.S.; Kim, E.; Kang, D.-H.; Choi, J.S.; Yu, K.-S.; Jang, I.-J.; Shin, S.-G. APLUS study group Taq1A Polymorphism in the Dopamine D2 Receptor Gene as a Predictor of Clinical Response to Aripiprazole. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2008, 18, 897–907. [Google Scholar] [CrossRef]
- Kim, E.; Kwon, J.S.; Shin, Y.-W.; Lee, J.S.; Kang, W.J.; Jo, H.J.; Lee, J.-M.; Yu, K.-S.; Kang, D.-H.; Cho, J.-Y.; et al. Taq1A Polymorphism in the Dopamine D2 Receptor Gene Predicts Brain Metabolic Response to Aripiprazole in Healthy Male Volunteers. Pharmacogenet. Genom. 2008, 18, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Pae, C.-U.; Chiesa, A.; Serretti, A. Influence of DAOA Gene Variants on Antipsychotic Response after Switch to Aripiprazole. Psychiatry Res. 2010, 178, 430–432. [Google Scholar] [CrossRef]
- Choong, E.; Polari, A.; Kamdem, R.H.; Gervasoni, N.; Spisla, C.; Jaquenoud Sirot, E.; Bickel, G.G.; Bondolfi, G.; Conus, P.; Eap, C.B. Pharmacogenetic Study on Risperidone Long-Acting Injection: Influence of Cytochrome P450 2D6 and Pregnane X Receptor on Risperidone Exposure and Drug-Induced Side-Effects. J. Clin. Psychopharmacol. 2013, 33, 289–298. [Google Scholar] [CrossRef]
- Wang, L.; Yu, L.; Zhang, A.-P.; Fang, C.; Du, J.; Gu, N.-F.; Qin, S.-Y.; Feng, G.-Y.; Li, X.-W.; Xing, Q.-H.; et al. Serum Prolactin Levels, Plasma Risperidone Levels, Polymorphism of Cytochrome P450 2D6 and Clinical Response in Patients with Schizophrenia. J. Psychopharmacol. Oxf. Engl. 2007, 21, 837–842. [Google Scholar] [CrossRef]
- Dutch Pharmacogenetics Working Group (DPWG) Risk Analysis Document for Risperidone and CYP2D6. Available online: https://www.g-standaard.nl/risicoanalyse/b0001536.pdf (accessed on 11 January 2023).
- Correia, C.T.; Almeida, J.P.; Santos, P.E.; Sequeira, A.F.; Marques, C.E.; Miguel, T.S.; Abreu, R.L.; Oliveira, G.G.; Vicente, A.M. Pharmacogenetics of Risperidone Therapy in Autism: Association Analysis of Eight Candidate Genes with Drug Efficacy and Adverse Drug Reactions. Pharm. J. 2010, 10, 418–430. [Google Scholar] [CrossRef]
- Ikeda, M.; Yamanouchi, Y.; Kinoshita, Y.; Kitajima, T.; Yoshimura, R.; Hashimoto, S.; O’Donovan, M.C.; Nakamura, J.; Ozaki, N.; Iwata, N. Variants of Dopamine and Serotonin Candidate Genes as Predictors of Response to Risperidone Treatment in First-Episode Schizophrenia. Pharmacogenomics 2008, 9, 1437–1443. [Google Scholar] [CrossRef]
- Xiong, Y.; Wei, Z.; Huo, R.; Wu, X.; Shen, L.; Li, Y.; Gong, X.; Wu, Z.; Feng, G.; Li, W.; et al. A Pharmacogenetic Study of Risperidone on Chemokine (C-C Motif) Ligand 2 (CCL2) in Chinese Han Schizophrenia Patients. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 51, 153–158. [Google Scholar] [CrossRef]
- Zhao, Q.-Z.; Liu, B.-C.; Zhang, J.; Wang, L.; Li, X.-W.; Wang, Y.; Ji, J.; Yang, F.-P.; Wan, C.-L.; Xu, Y.-F.; et al. Association between a COMT Polymorphism and Clinical Response to Risperidone Treatment: A Pharmacogenetic Study. Psychiatr. Genet. 2012, 22, 298–299. [Google Scholar] [CrossRef]
- Urichuk, L.; Prior, T.I.; Dursun, S.; Baker, G. Metabolism of Atypical Antipsychotics: Involvement of Cytochrome P450 Enzymes and Relevance for Drug-Drug Interactions. Curr. Drug Metab. 2008, 9, 410–418. [Google Scholar] [CrossRef]
- Zubiaur, P.; Soria-Chacartegui, P.; Villapalos-García, G.; Gordillo-Perdomo, J.J.; Abad-Santos, F. The Pharmacogenetics of Treatment with Olanzapine. Pharmacogenomics 2021, 22, 939–958. [Google Scholar] [CrossRef] [PubMed]
- Ghotbi, R.; Mannheimer, B.; Aklillu, E.; Suda, A.; Bertilsson, L.; Eliasson, E.; Osby, U. Carriers of the UGT1A4 142T>G Gene Variant Are Predisposed to Reduced Olanzapine Exposure–an Impact Similar to Male Gender or Smoking in Schizophrenic Patients. Eur. J. Clin. Pharmacol. 2010, 66, 465–474. [Google Scholar] [CrossRef]
- Young, R.M.; Lawford, B.R.; Barnes, M.; Burton, S.C.; Ritchie, T.; Ward, W.K.; Noble, E.P. Prolactin Levels in Antipsychotic Treatment of Patients with Schizophrenia Carrying the DRD2*A1 Allele. Br. J. Psychiatry J. Ment. Sci. 2004, 185, 147–151. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.-J.; Liou, Y.-J.; Bai, Y.M.; Chen, T.-T.; Wang, Y.-C.; Tsai, S.-J. Dopamine Receptor D2 Gene Is Associated with Weight Gain in Schizophrenic Patients under Long-Term Atypical Antipsychotic Treatment. Pharmacogenet. Genom. 2010, 20, 359–366. [Google Scholar] [CrossRef]
- Tiwari, A.K.; Zai, C.C.; Likhodi, O.; Lisker, A.; Singh, D.; Souza, R.P.; Batra, P.; Zaidi, S.H.E.; Chen, S.; Liu, F.; et al. A Common Polymorphism in the Cannabinoid Receptor 1 (CNR1) Gene Is Associated with Antipsychotic-Induced Weight Gain in Schizophrenia. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2010, 35, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Calarge, C.A.; Ellingrod, V.L.; Zimmerman, B.; Acion, L.; Sivitz, W.I.; Schlechte, J.A. Leptin Gene -2548G/A Variants Predict Risperidone-Associated Weight Gain in Children and Adolescents. Psychiatr. Genet. 2009, 19, 320–327. [Google Scholar] [CrossRef]
- Koller, D.; Almenara, S.; Mejía, G.; Saiz-Rodríguez, M.; Zubiaur, P.; Román, M.; Ochoa, D.; Navares-Gómez, M.; Santos-Molina, E.; Pintos-Sánchez, E.; et al. Metabolic Effects of Aripiprazole and Olanzapine Multiple-Dose Treatment in a Randomised Crossover Clinical Trial in Healthy Volunteers: Association with Pharmacogenetics. Adv. Ther. 2021, 38, 1035–1054. [Google Scholar] [CrossRef]
- Wheelock, V. The Motor Disorder. In A Physician’s Guide to the Management of Huntington’s Disease, 3rd ed.; Nance, M., Paulsen, J.S., Rosenblatt, A., Wheelock, V., Eds.; Huntington’s Disease Society of America: New York, NY, USA, 2011. [Google Scholar]
- Kato, Y.; Nakajima, M.; Oda, S.; Fukami, T.; Yokoi, T. Human UDP-Glucuronosyltransferase Isoforms Involved in Haloperidol Glucuronidation and Quantitative Estimation of Their Contribution. Drug Metab. Dispos. Biol. Fate Chem. 2012, 40, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Kudo, S.; Ishizaki, T. Pharmacokinetics of Haloperidol: An Update. Clin. Pharmacokinet. 1999, 37, 435–456. [Google Scholar] [CrossRef]
- Murray, M. Role of CYP Pharmacogenetics and Drug-Drug Interactions in the Efficacy and Safety of Atypical and Other Antipsychotic Agents. J. Pharm. Pharmacol. 2006, 58, 871–885. [Google Scholar] [CrossRef]
- Ohara, K.; Tanabu, S.; Yoshida, K.; Ishibashi, K.; Ikemoto, K.; Shibuya, H. Effects of Smoking and Cytochrome P450 2D6*10 Allele on the Plasma Haloperidol Concentration/Dose Ratio. Prog. Neuropsychopharmacol. Biol. Psychiatry 2003, 27, 945–949. [Google Scholar] [CrossRef]
- Someya, T.; Shimoda, K.; Suzuki, Y.; Sato, S.; Kawashima, Y.; Hirokane, G.; Morita, S.; Yokono, A.; Takahashi, S. Effect of CYP2D6 Genotypes on the Metabolism of Haloperidol in a Japanese Psychiatric Population. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2003, 28, 1501–1505. [Google Scholar] [CrossRef]
- Dutch Pharmacogenetics Working Group (DPWG) Risk Assessment. Available online: https://api.pharmgkb.org/v1/download/file/attachment/dpwg_may_2021.pdf (accessed on 11 January 2023).
- Fukasawa, T.; Suzuki, A.; Otani, K. Effects of Genetic Polymorphism of Cytochrome P450 Enzymes on the Pharmacokinetics of Benzodiazepines. J. Clin. Pharm. Ther. 2007, 32, 333–341. [Google Scholar] [CrossRef]
- Zubiaur, P.; Abad-Santos, F. Use of Pharmacogenetics for Benzodiazepine Prescription: State of the Art and Expectations. Pharmacogenomics 2022, 23, 949–952. [Google Scholar] [CrossRef]
- Epping, E.A.; Kim, J.-I.; Craufurd, D.; Brashers-Krug, T.M.; Anderson, K.E.; McCusker, E.; Luther, J.; Long, J.D.; Paulsen, J.S. PREDICT-HD Investigators and Coordinators of the Huntington Study Group Longitudinal Psychiatric Symptoms in Prodromal Huntington’s Disease: A Decade of Data. Am. J. Psychiatry 2016, 173, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Paoli, R.; Botturi, A.; Ciammola, A.; Silani, V.; Prunas, C.; Lucchiari, C.; Zugno, E.; Caletti, E. Neuropsychiatric Burden in Huntington’s Disease. Brain Sci. 2017, 7, 67. [Google Scholar] [CrossRef]
- Paulsen, J.S. Neuropsychiatric Aspects of Huntington’s Disease. J. Neurol. Neurosurg. Psychiatry 2001, 71, 310–314. [Google Scholar] [CrossRef] [Green Version]
- Duff, K.; Paulsen, J.S.; Beglinger, L.J.; Langbehn, D.R.; Stout, J.C. Psychiatric Symptoms in Huntington’s Disease before Diagnosis: The Predict-HD Study. Biol. Psychiatry 2007, 62, 1341–1346. [Google Scholar] [CrossRef]
- Goh, A.M.; Wibawa, P.; Loi, S.M.; Walterfang, M.; Velakoulis, D.; Looi, J.C. Huntington’s Disease: Neuropsychiatric Manifestations of Huntington’s Disease. Australas. Psychiatry 2018, 26, 366–375. [Google Scholar] [CrossRef]
- Loi, S.M.; Walterfang, M.; Velakoulis, D.; Looi, J.C. Huntington’s Disease: Managing Neuropsychiatric Symptoms in Huntington’s Disease. Australas. Psychiatry 2018, 26, 376–380. [Google Scholar] [CrossRef]
- Beglinger, L.J.; Adams, W.H.; Langbehn, D.; Fiedorowicz, J.G.; Jorge, R.; Biglan, K.; Caviness, J.; Olson, B.; Robinson, R.G.; Kieburtz, K.; et al. Results of the Citalopram to Enhance Cognition in Huntington Disease Trial: Citalopram in HD. Mov. Disord. 2014, 29, 401–405. [Google Scholar] [CrossRef] [Green Version]
- Hicks, J.K.; Bishop, J.R.; Sangkuhl, K.; Müller, D.J.; Ji, Y.; Leckband, S.G.; Leeder, J.S.; Graham, R.L.; Chiulli, D.L.; LLerena, A.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6 and CYP2C19 Genotypes and Dosing of Selective Serotonin Reuptake Inhibitors. Clin. Pharmacol. Ther. 2015, 98, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Klamerus, K.J.; Maloney, K.; Rudolph, R.L.; Sisenwine, S.F.; Jusko, W.J.; Chiang, S.T. Introduction of a Composite Parameter to the Pharmacokinetics of Venlafaxine and Its Active O-Desmethyl Metabolite. J. Clin. Pharmacol. 1992, 32, 716–724. [Google Scholar] [CrossRef]
- Sangkuhl, K.; Stingl, J.C.; Turpeinen, M.; Altman, R.B.; Klein, T.E. PharmGKB Summary: Venlafaxine Pathway. Pharmacogenet. Genom. 2014, 24, 62–72. [Google Scholar] [CrossRef] [Green Version]
- Otton, S.V.; Ball, S.E.; Cheung, S.W.; Inaba, T.; Rudolph, R.L.; Sellers, E.M. Venlafaxine Oxidation in Vitro Is Catalysed by CYP2D6. Br. J. Clin. Pharmacol. 1996, 41, 149–156. [Google Scholar] [CrossRef]
- Fogelman, S.M.; Schmider, J.; Venkatakrishnan, K.; von Moltke, L.L.; Harmatz, J.S.; Shader, R.I.; Greenblatt, D.J. O- and N-Demethylation of Venlafaxine in Vitro by Human Liver Microsomes and by Microsomes from CDNA-Transfected Cells: Effect of Metabolic Inhibitors and SSRI Antidepressants. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 1999, 20, 480–490. [Google Scholar] [CrossRef]
- Lobello, K.W.; Preskorn, S.H.; Guico-Pabia, C.J.; Jiang, Q.; Paul, J.; Nichols, A.I.; Patroneva, A.; Ninan, P.T. Cytochrome P450 2D6 Phenotype Predicts Antidepressant Efficacy of Venlafaxine: A Secondary Analysis of 4 Studies in Major Depressive Disorder. J. Clin. Psychiatry 2010, 71, 1482–1487. [Google Scholar] [CrossRef]
- Nichols, A.I.; Focht, K.; Jiang, Q.; Preskorn, S.H.; Kane, C.P. Pharmacokinetics of Venlafaxine Extended Release 75 Mg and Desvenlafaxine 50 Mg in Healthy CYP2D6 Extensive and Poor Metabolizers: A Randomized, Open-Label, Two-Period, Parallel-Group, Crossover Study. Clin. Drug Investig. 2011, 31, 155–167. [Google Scholar] [CrossRef]
- McAlpine, D.E.; Biernacka, J.M.; Mrazek, D.A.; O’Kane, D.J.; Stevens, S.R.; Langman, L.J.; Courson, V.L.; Bhagia, J.; Moyer, T.P. Effect of Cytochrome P450 Enzyme Polymorphisms on Pharmacokinetics of Venlafaxine. Ther. Drug Monit. 2011, 33, 14–20. [Google Scholar] [CrossRef]
- Dutch Pharmacogenetics Working Group. Annotation of DPWG Guideline for Venlafaxine and CYP2D6. Available online: https://www.pharmgkb.org/chemical/pa451866/guidelineannotation/pa166104968 (accessed on 24 January 2023).
- Fukuda, T.; Nishida, Y.; Zhou, Q.; Yamamoto, I.; Kondo, S.; Azuma, J. The Impact of the CYP2D6 and CYP2C19 Genotypes on Venlafaxine Pharmacokinetics in a Japanese Population. Eur. J. Clin. Pharmacol. 2000, 56, 175–180. [Google Scholar] [CrossRef]
- Uhr, M.; Tontsch, A.; Namendorf, C.; Ripke, S.; Lucae, S.; Ising, M.; Dose, T.; Ebinger, M.; Rosenhagen, M.; Kohli, M.; et al. Polymorphisms in the Drug Transporter Gene ABCB1 Predict Antidepressant Treatment Response in Depression. Neuron 2008, 57, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Lohoff, F.W.; Aquino, T.D.; Narasimhan, S.; Multani, P.K.; Etemad, B.; Rickels, K. Serotonin Receptor 2A (HTR2A) Gene Polymorphism Predicts Treatment Response to Venlafaxine XR in Generalized Anxiety Disorder. Pharm. J. 2013, 13, 21–26. [Google Scholar] [CrossRef] [Green Version]
- Kirchheiner, J.; Nickchen, K.; Sasse, J.; Bauer, M.; Roots, I.; Brockmöller, J. A 40-Basepair VNTR Polymorphism in the Dopamine Transporter (DAT1) Gene and the Rapid Response to Antidepressant Treatment. Pharm. J. 2007, 7, 48–55. [Google Scholar] [CrossRef] [Green Version]
- U.S. Food and Drug Administration (FDA) Label Information for Duloxetine. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/022516lbl.pdf (accessed on 16 January 2023).
- Knadler, M.P.; Lobo, E.; Chappell, J.; Bergstrom, R. Duloxetine: Clinical Pharmacokinetics and Drug Interactions. Clin. Pharmacokinet. 2011, 50, 281–294. [Google Scholar] [CrossRef]
- Anttila, S.A.K.; Leinonen, E.V.J. A Review of the Pharmacological and Clinical Profile of Mirtazapine. CNS Drug Rev. 2006, 7, 249–264. [Google Scholar] [CrossRef]
- Lind, A.-B.; Reis, M.; Bengtsson, F.; Jonzier-Perey, M.; Powell Golay, K.; Ahlner, J.; Baumann, P.; Dahl, M.-L. Steady-State Concentrations of Mirtazapine, N-Desmethylmirtazapine, 8-Hydroxymirtazapine and Their Enantiomers in Relation to Cytochrome P450 2D6 Genotype, Age and Smoking Behaviour. Clin. Pharmacokinet. 2009, 48, 63–70. [Google Scholar] [CrossRef]
- Sirot, E.J.; Harenberg, S.; Vandel, P.; Lima, C.A.M.; Perrenoud, P.; Kemmerling, K.; Zullino, D.F.; Hilleret, H.; Crettol, S.; Jonzier-Perey, M.; et al. Multicenter Study on the Clinical Effectiveness, Pharmacokinetics, and Pharmacogenetics of Mirtazapine in Depression. J. Clin. Psychopharmacol. 2012, 32, 622–629. [Google Scholar] [CrossRef]
- Levy, R.; Czernecki, V. Apathy and the Basal Ganglia. J. Neurol. 2006, 253, vii54–vii61. [Google Scholar] [CrossRef]
- Bouwens, J.A.; van Duijn, E.; van der Mast, R.C.; Roos, R.A.C.; Giltay, E.J. Irritability in a Prospective Cohort of Huntington’s Disease Mutation Carriers. J. Neuropsychiatry Clin. Neurosci. 2015, 27, 206–212. [Google Scholar] [CrossRef]
- U.S. Food & Drug Administration (FDA) Label Information for Clozapine. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/019758s062lbl.pdf (accessed on 17 January 2023).
- Saiz-Rodríguez, M.; Gil-Polo, C.; Diez-Fairen, M.; Martinez-Horta, S.-I.; Sampedro Santalo, F.; Collazo, C.; Calvo, S.; Alonso-García, E.; Riñones-Mena, E.; Aguado, L.; et al. Polymorphisms in the Oxytocin Receptor and Their Association with Apathy and Impaired Social Cognition in Huntington’s Disease. J. Neurol. Neurosurg. Psychiatry 2022, 93, A50. [Google Scholar] [CrossRef]
- Wesson, M.; Boileau, N.R.; Perlmutter, J.S.; Paulsen, J.S.; Barton, S.K.; McCormack, M.K.; Carlozzi, N.E. Suicidal Ideation Assessment in Individuals with Premanifest and Manifest Huntington Disease. J. Huntingt. Dis. 2018, 7, 239–249. [Google Scholar] [CrossRef]


{kind=link}
| Phenotype * | Paroxetine | Fluvoxamine | Citalopram | Escitalopram | Sertraline |
|---|---|---|---|---|---|
| UM | Consider alternative drug not predominantly metabolized by CYP2D6. | No recommendation. | Consider an alternative drug not predominantly metabolized by CYP2C19. | Consider an alternative drug not predominantly metabolized by CYP2C19. | If patient does not respond to recommended starting dose, consider alternative drug not predominantly metabolized by CYP2C19. |
| NM | Initiate therapy with recommended starting dose. | Initiate therapy with recommended starting dose. | Initiate therapy with recommended starting dose. | Initiate therapy with recommended starting dose. | Initiate therapy with recommended starting dose. |
| IM | Initiate therapy with recommended starting dose. | Initiate therapy with recommended starting dose. | Initiate therapy with recommended starting dose. | Initiate therapy with recommended starting dose. | Initiate therapy with recommended starting dose. |
| PM | Consider alternative drug not predominantly metabolized by CYP2D6 or consider a 50% reduction of recommended starting dose. | Consider alternative drug not predominantly metabolized by CYP2D6 or consider a 25–50% reduction of recommended starting dose. | Consider alternative drug not predominantly metabolized by CYP2C19 or consider a 50% reduction of recommended starting dose. | Consider alternative drug not predominantly metabolized by CYP2C19 or consider a 50% reduction of recommended starting dose. | Consider alternative drug not predominantly metabolized by CYP2C19 or consider a 50% reduction of recommended starting dose. |
| CYP2D6 * | CYP2C19 * | ||||
| Gene | Drug(s) Related | HD Symptom |
|---|---|---|
| ABCB1 | Risperidone | Chorea |
| Psychiatric symptoms | ||
| Anxiety | ||
| Olanzapine | Chorea | |
| Psychiatric symptoms | ||
| Anxiety | ||
| Venlafaxine | Depression | |
| Anxiety | ||
| AKT1 | Risperidone | Chorea |
| Psychiatric symptoms | ||
| Anxiety | ||
| ANKK1 | Aripiprazole | Chorea |
| Psychiatric symptoms | ||
| Anxiety | ||
| BDNF | Venlafaxine | Depression |
| Anxiety | ||
| CCL2 | Risperidone | Chorea |
| Psychiatric symptoms | ||
| Anxiety | ||
| COMT | Risperidone | Chorea |
| Psychiatric symptoms | ||
| Anxiety | ||
| Venlafaxine | Depression | |
| Anxiety | ||
| CYP1A2 | Olanzapine | Chorea |
| Psychiatric symptoms | ||
| Anxiety | ||
| Duloxetine | Depression | |
| Anxiety | ||
| Clozapine | Chorea | |
| Psychiatric symptoms | ||
| Anxiety | ||
| Psychosis | ||
| Mirtazapine | Depression | |
| CYP2C19 | Escitalopram | Depression |
| Irritability | ||
| Apathy | ||
| Anxiety | ||
| Citalopram | Depression | |
| Irritability | ||
| Apathy | ||
| Anxiety | ||
| Sertraline | Depression | |
| Irritability | ||
| Apathy | ||
| Anxiety | ||
| Venlafaxine | Depression | |
| Anxiety | ||
| Diazepam | Agitation and anxiety | |
| CYP2D6 | Tetrabenazine | Chorea |
| Deutetrabenazine | ||
| Aripiprazole | Chorea | |
| Psychiatric symptoms | ||
| Anxiety | ||
| Psychosis | ||
| Risperidone | Chorea | |
| Psychiatric symptoms | ||
| Anxiety | ||
| Psychosis | ||
| Olanzapine | Chorea | |
| Psychiatric symptoms | ||
| Anxiety | ||
| Psychosis | ||
| Clozapine | Chorea | |
| Psychiatric symptoms | ||
| Anxiety | ||
| Psychosis | ||
| Haloperidol | Chorea | |
| Psychiatric symptoms | ||
| Irritability | ||
| Anxiety | ||
| Psychosis | ||
| Paroxetine | Depression | |
| Irritability | ||
| Apathy | ||
| Anxiety | ||
| Fluvoxamine | Depression | |
| Irritability | ||
| Apathy | ||
| Anxiety | ||
| Venlafaxine | Depression | |
| Anxiety | ||
| Duloxetine | Depression | |
| Anxiety | ||
| Mirtazapine | Depression | |
| Irritability | ||
| CYP3A4 | Aripiprazole | Chorea |
| Psychiatric symptoms and anxiety | ||
| Risperidone | Chorea | |
| Psychiatric symptoms | ||
| Anxiety | ||
| Olanzapine | Chorea | |
| Psychiatric symptoms | ||
| Anxiety | ||
| Clozapine | Chorea | |
| Psychiatric symptoms | ||
| Anxiety | ||
| Psychosis | ||
| Haloperidol | Chorea | |
| Psychiatric symptoms | ||
| Anxiety | ||
| Mirtazapine | Depression | |
| Diazepam | Agitation and anxiety | |
| Clobazam | Agitation and anxiety | |
| Midazolam | Agitation and anxiety | |
| Clonazepam | Agitation and anxiety | |
| Alprazolam | Agitation and anxiety | |
| Tiazolam | Agitation and anxiety | |
| DAOA | Aripiprazole | Chorea |
| Psychiatric symptoms | ||
| Anxiety | ||
| DRD2 | Aripiprazole | Chorea |
| Psychiatric symptoms | ||
| Anxiety | ||
| Olanzapine | Chorea | |
| Psychiatric symptoms | ||
| Anxiety | ||
| DRD3 | Olanzapine | Chorea |
| Psychiatric symptoms | ||
| Anxiety | ||
| HTR2A | Olanzapine | Chorea |
| Psychiatric symptoms | ||
| Anxiety | ||
| Venlafaxine | Depression | |
| Anxiety | ||
| LEP | Olanzapine | Chorea |
| Psychiatric symptoms | ||
| Anxiety | ||
| OXTR | No drug—related | Apathy |
| Irritability | ||
| Impaired social cognition | ||
| SLC6A4 | Venlafaxine | Depression |
| Anxiety | ||
| UGT1A4 | Haloperidol | Chorea |
| Psychiatric symptoms | ||
| Anxiety | ||
| UGT1A9 | Haloperidol | Chorea |
| Psychiatric symptoms | ||
| Anxiety | ||
| UGT2B7 | Haloperidol | Chorea |
| Psychiatric symptoms | ||
| Anxiety |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-González, X.; Cubo, E.; Simón-Vicente, L.; Mariscal, N.; Alcaraz, R.; Aguado, L.; Rivadeneyra-Posadas, J.; Sanz-Solas, A.; Saiz-Rodríguez, M. Pharmacogenetics in the Treatment of Huntington’s Disease: Review and Future Perspectives. J. Pers. Med. 2023, 13, 385. https://doi.org/10.3390/jpm13030385
García-González X, Cubo E, Simón-Vicente L, Mariscal N, Alcaraz R, Aguado L, Rivadeneyra-Posadas J, Sanz-Solas A, Saiz-Rodríguez M. Pharmacogenetics in the Treatment of Huntington’s Disease: Review and Future Perspectives. Journal of Personalized Medicine. 2023; 13(3):385. https://doi.org/10.3390/jpm13030385
Chicago/Turabian StyleGarcía-González, Xandra, Esther Cubo, Lucía Simón-Vicente, Natividad Mariscal, Raquel Alcaraz, Laura Aguado, Jéssica Rivadeneyra-Posadas, Antonio Sanz-Solas, and Miriam Saiz-Rodríguez. 2023. "Pharmacogenetics in the Treatment of Huntington’s Disease: Review and Future Perspectives" Journal of Personalized Medicine 13, no. 3: 385. https://doi.org/10.3390/jpm13030385