
Bone Metastasis in Bladder Cancer


{kind=link}
Abstract
:1. Introduction
2. Epidemiology
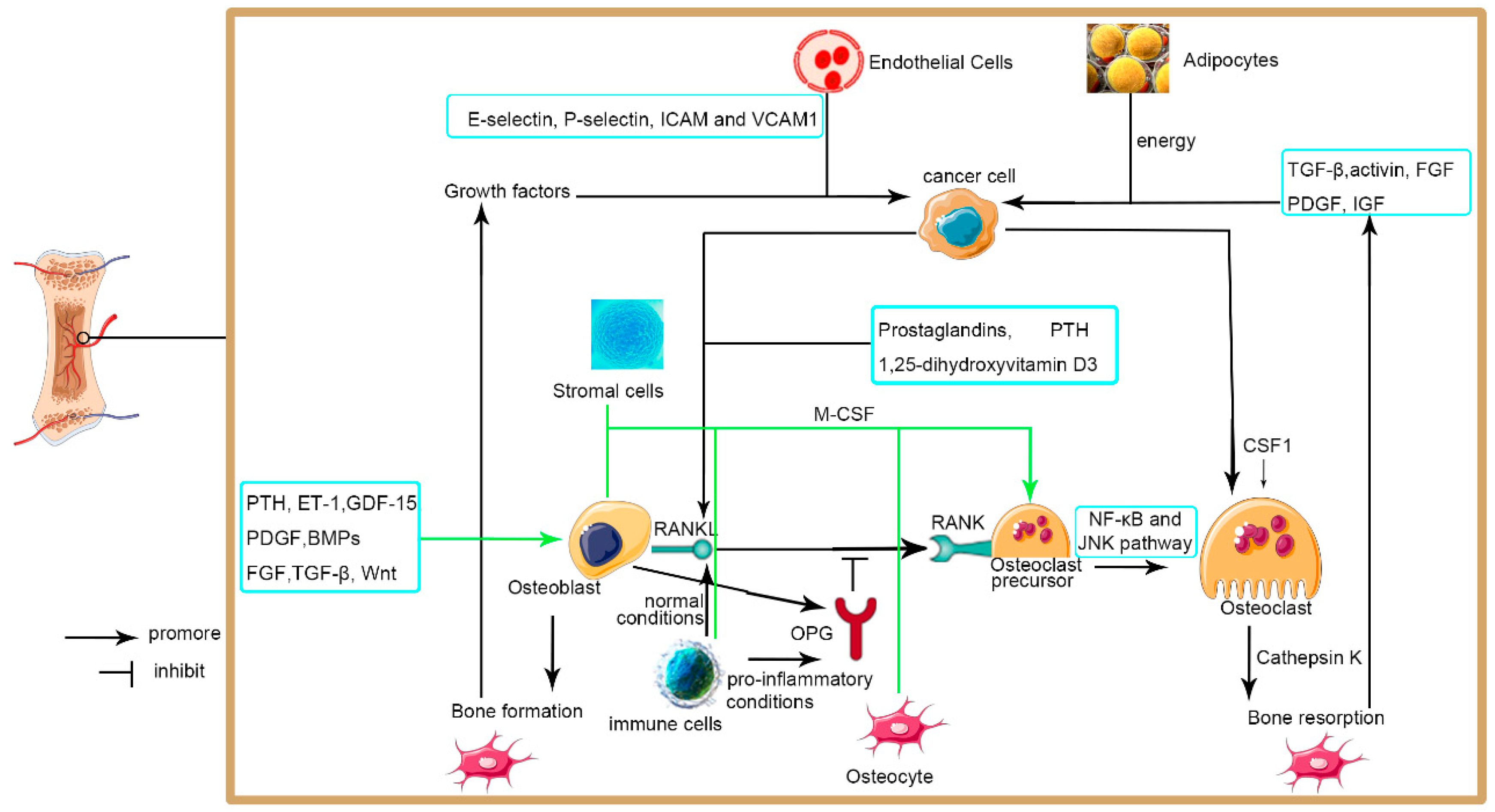
3. Mechanisms/Pathophysiology
3.1. Epithelial-to-Mesenchymal Transition
3.2. Angiogenesis, Intravasation and Extravasation
3.3. Metastases to Bone
3.3.1. Bone Microenvironment
3.3.2. Osteoclasts
3.3.3. Osteoblasts
3.3.4. Endothelial Cells
3.3.5. Immune Cells
3.3.6. Adipocytes
3.3.7. Osteolytic Metastasis
3.3.8. Osteoblastic Metastasis
3.4. Other Mechanisms
4. Preclinical Model
5. Diagnosis
5.1. Skeletal-Related Events (SREs)
5.2. Imaging
5.3. Biopsy
5.4. Biochemical Markers
6. Management
6.1. Chemotherapy
6.1.1. First-Line Chemotherapy for Metastatic Disease
6.1.2. Second-Line Chemotherapy and beyond for Metastatic Disease
6.2. Targeted Therapies and Antibody-Drug Conjugates
6.3. Bisphosphonates
6.4. Denosumab
6.5. Clinical Use of Bisphosphonates and Denosumab
6.6. Radioisotopes
6.7. External Beam Radiotherapy
6.8. Surgery
6.9. Therapeutic Strategies of Bone Pain
7. Conclusions and Future Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Kamat, A.M.; Hahn, N.M.; Efstathiou, J.A.; Lerner, S.P.; Malmström, P.-U.; Choi, W.; Guo, C.C.; Lotan, Y.; Kassouf, W. Bladder cancer. Lancet 2016, 388, 2796–2810. [Google Scholar] [CrossRef] [PubMed]
- Witjes, J.A.; Bruins, H.M.; Cathomas, R.; Compérat, E.M.; Cowan, N.C.; Gakis, G.; Hernández, V.; Espinós, E.L.; Lorch, A.; Neuzillet, Y.; et al. European Association of Urology Guidelines on Muscle-invasive and Metastatic Bladder Cancer: Summary of the 2020 Guidelines. Eur. Urol. 2020, 79, 82–104. [Google Scholar] [CrossRef] [PubMed]
- Witjes, J.A.; Compérat, E.; Cowan, N.C.; De Santis, M.; Gakis, G.; Lebret, T.; Ribal, M.J.; Van der Heijden, A.G.; Sherif, A. EAU Guidelines on Muscle-invasive and Metastatic Bladder Cancer: Summary of the 2013 Guidelines. Eur. Urol. 2013, 65, 778–792. [Google Scholar] [CrossRef]
- Shinagare, A.B.; Ramaiya, N.H.; Jagannathan, J.P.; Fennessy, F.M.; Taplin, M.-E.; Abbeele, A.D.V.D. Metastatic Pattern of Bladder Cancer: Correlation With the Characteristics of the Primary Tumor. Am. J. Roentgenol. 2011, 196, 117–122. [Google Scholar] [CrossRef]
- Coleman, R. Metastatic bone disease: Clinical features, pathophysiology and treatment strategies. Cancer Treat. Rev. 2001, 27, 165–176. [Google Scholar] [CrossRef]
- Fan, Z.; Huang, Z.; Hu, C.; Tong, Y.; Zhao, C. Risk factors and nomogram for newly diagnosis of bone metastasis in bladder cancer. Medicine 2020, 99, e22675. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, L.; Tao, F.; Guo, X.; Feng, G.; Chen, F.; Xu, Y.; Li, L.; Han, X.; Baklaushev, V.P.; et al. Bone Metastases Pattern in Newly Diagnosed Metastatic Bladder Cancer: A Population-Based Study. J. Cancer 2018, 9, 4706–4711. [Google Scholar] [CrossRef]
- Svensson, E.; Christiansen, C.; Ulrichsen, S.P.; Rørth, M.R.; Sørensen, H.T. Survival after bone metastasis by primary cancer type: A Danish population-based cohort study. BMJ Open 2017, 7, e016022. [Google Scholar] [CrossRef]
- Tsuda, Y.; Nakagawa, T.; Shinoda, Y.; Kanatani, A.; Kawai, T.; Taguchi, S.; Yamada, Y.; Sawada, R.; Kume, H.; Homma, Y.; et al. Skeletal-related events and prognosis in urothelial cancer patients with bone metastasis. Int. J. Clin. Oncol. 2017, 22, 548–553. [Google Scholar] [CrossRef]
- Von Der Maase, H.; Sengelov, L.; Roberts, J.T.; Ricci, S.; Dogliotti, L.; Oliver, T.; Moore, M.J.; Zimmermann, A.; Arning, M. Long-Term Survival Results of a Randomized Trial Comparing Gemcitabine Plus Cisplatin, With Methotrexate, Vinblastine, Doxorubicin, Plus Cisplatin in Patients With Bladder Cancer. J. Clin. Oncol. 2005, 23, 4602–4608. [Google Scholar] [CrossRef] [PubMed]
- Batson, O.V. The Function of the Vertebral Veins and Their Role in the Spread of Metastases. Ann. Surg. 1940, 112, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Foschi, N.; Ragonese, M.; Grassi, V.M.; De Matteis, V.; De-Giorgio, F. The periprostatic venous plexus: An unusual source of fatal pulmonary embolism during corporoplasty. Int. J. Leg. Med. 2016, 131, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Nathoo, N.; Caris, E.C.; Wiener, J.A.; Mendel, E. History of the Vertebral Venous Plexus and the Significant Contributions of Breschet and Batson. Neurosurgery 2011, 69, 1007–1014. [Google Scholar] [CrossRef] [Green Version]
- Seton-Rogers, S. Teaching old macrophages new tricks. Nat. Rev. Cancer 2013, 13, 753. [Google Scholar] [CrossRef]
- Qian, B.-Z.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Psaila, B.; Lyden, D. The metastatic niche: Adapting the foreign soil. Nat. Rev. Cancer 2009, 9, 285–293. [Google Scholar] [CrossRef]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2008, 9, 239–252. [Google Scholar] [CrossRef]
- Fidler, I.J. Critical determinants of metastasis. Semin. Cancer Biol. 2002, 12, 89–96. [Google Scholar] [CrossRef]
- Coniglio, S.J. Role of Tumor-Derived Chemokines in Osteolytic Bone Metastasis. Front. Endocrinol. 2018, 9, 313. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Brennan, J.P.; Slavin, J.L.; Blick, T.; Thompson, E.W.; Williams, E.D. Mesenchymal-to-Epithelial Transition Facilitates Bladder Cancer Metastasis: Role of Fibroblast Growth Factor Receptor-2. Cancer Res. 2006, 66, 11271–11278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Qi, L.; Liu, S.; Liu, W.; Ou, Z.; Chen, M.; Liu, L.; Zu, X.; Wang, J.; Li, Y. CLASP2 is involved in the EMT and early progression after transurethral resection of the bladder tumor. BMC Cancer 2017, 17, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.-K.; Dayyani, F.; Gallick, G.E. Steps in prostate cancer progression that lead to bone metastasis. Int. J. Cancer 2011, 128, 2545–2561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Roy, F.; Berx, G. The cell-cell adhesion molecule E-cadherin. Cell. Mol. Life Sci. 2008, 65, 3756–3788. [Google Scholar] [CrossRef] [PubMed]
- Takeichi, M. Cadherin Cell Adhesion Receptors as a Morphogenetic Regulator. Science 1991, 251, 1451–1455. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial–mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Matsumoto, R.; Tsuda, M.; Wang, L.; Maishi, N.; Abe, T.; Kimura, T.; Tanino, M.; Nishihara, H.; Hida, K.; Ohba, Y.; et al. Adaptor protein CRK induces epithelial–mesenchymal transition and metastasis of bladder cancer cells through HGF/c-Met feedback loop. Cancer Sci. 2015, 106, 709–717. [Google Scholar] [CrossRef]
- Long, X.; Xiong, W.; Zeng, X.; Qi, L.; Cai, Y.; Mo, M.; Jiang, H.; Zhu, B.; Chen, Z.; Li, Y. Cancer-associated fibroblasts promote cisplatin resistance in bladder cancer cells by increasing IGF-1/ERβ/Bcl-2 signalling. Cell Death Dis. 2019, 10, 375. [Google Scholar] [CrossRef] [Green Version]
- Gdowski, A.S.; Ranjan, A.; Vishwanatha, J.K. Current concepts in bone metastasis, contemporary therapeutic strategies and ongoing clinical trials. J. Exp. Clin. Cancer Res. 2017, 36, 108. [Google Scholar] [CrossRef]
- DU, Z.; Hou, S. The Anti-Angiogenic Activity of Human Endostatin Inhibits Bladder Cancer Growth and Its Mechanism. J. Urol. 2003, 170, 2000–2003. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, Y.; Su, L.-J.; Flaig, T.W. VEGFR and EGFR inhibition increases epithelial cellular characteristics and chemotherapy sensitivity in mesenchymal bladder cancer cells. Oncol. Rep. 2010, 24, 1019–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weis, S.M.; Cheresh, D.A. Tumor angiogenesis: Molecular pathways and therapeutic targets. Nat. Med. 2011, 17, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Reymond, N.; Im, J.H.; Garg, R.; Vega, F.M.; D’Agua, B.B.; Riou, P.; Cox, S.; Valderrama, F.; Muschel, R.J.; Ridley, A.J. Cdc42 promotes transendothelial migration of cancer cells through β1 integrin. J. Cell Biol. 2012, 199, 653–668. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.P. Cell adhesion molecules of the immunoglobulin supergene family and their role in malignant transformation and progression to metastatic disease. Cancer Metast. Rev. 1991, 10, 11–22. [Google Scholar] [CrossRef]
- Inoue, K.; Karashima, T.; Fukata, S.; Nomura, A.; Kawada, C.; Kurabayashi, A.; Furihata, M.; Ohtsuki, Y.; Shuin, T. Effect of Combination Therapy with a Novel Bisphosphonate, Minodronate (YM529), and Docetaxel on a Model of Bone Metastasis by Human Transitional Cell Carcinoma. Clin. Cancer Res. 2005, 11, 6669–6677. [Google Scholar] [CrossRef] [Green Version]
- Ahmadzadeh, A.; Kast, R.E.; Ketabchi, N.; Shahrabi, S.; Shahjahani, M.; Jaseb, K.; Saki, N. Regulatory effect of chemokines in bone marrow niche. Cell Tissue Res. 2015, 361, 401–410. [Google Scholar] [CrossRef]
- Tencerova, M.; Kassem, M. The Bone Marrow-Derived Stromal Cells: Commitment and Regulation of Adipogenesis. Front. Endocrinol. 2016, 7, 127. [Google Scholar] [CrossRef] [Green Version]
- Roodman, G.D. Cell biology of the osteoclast. Exp. Hematol. 1999, 27, 1229–1241. [Google Scholar] [CrossRef]
- Kodama, H.; Nose, M.; Niida, S.; Yamasaki, A. Essential role of macrophage colony-stimulating factor in the osteoclast differentiation supported by stromal cells. J. Exp. Med. 1991, 173, 1291–1294. [Google Scholar] [CrossRef]
- Roodman, G.D. Mechanisms of Bone Metastasis. N. Engl. J. Med. 2004, 350, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Simonet, W.S.; Lacey, D.L.; Dunstan, C.R.; Kelley, M.; Chang, M.S.; Luthy, R.; Nguyen, H.Q.; Wooden, S.; Bennett, L.; Boone, T.; et al. Osteoprotegerin: A Novel Secreted Protein Involved in the Regulation of Bone Density. Cell 1997, 89, 309–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, H.; Morony, S.; Sarosi, I.; Dunstan, C.R.; Capparelli, C.; Scully, S.; Van, G.; Kaufman, S.; Kostenuik, P.J.; Lacey, D.L.; et al. Osteoprotegerin Reverses Osteoporosis by Inhibiting Endosteal Osteoclasts and Prevents Vascular Calcification by Blocking a Process Resembling Osteoclastogenesis. J. Exp. Med. 2000, 192, 463–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubin, J.E. Bone stem cells. J. Cell. Biochem. 1998, 72 (Suppl. 30–31), 73–82. [Google Scholar] [CrossRef]
- Wozney, J.M. Overview of Bone Morphogenetic Proteins. Spine 2002, 27, S2–S8. [Google Scholar] [CrossRef]
- Mundy, G.R.; Chen, D.; Zhao, M.; Dallas, S.; Xu, C.; Harris, S. Growth regulatory factors and bone. Rev. Endocr. Metab. Disord. 2001, 2, 105–115. [Google Scholar] [CrossRef]
- Stein, G.S.; Lian, J.B. Molecular Mechanisms Mediating Proliferation/Differentiation Interrelationships during Progressive Development of the Osteoblast Phenotype. Endocr. Rev. 1993, 14, 424–442. [Google Scholar] [CrossRef]
- Glinsky, V.V. Intravascular cell-to-cell adhesive interactions and bone metastasis. Cancer Metast. Rev. 2006, 25, 531–540. [Google Scholar] [CrossRef]
- Cook, L.M.; Shay, G.; Aruajo, A.; Lynch, C.C. Integrating new discoveries into the “vicious cycle” paradigm of prostate to bone metastases. Cancer Metast. Rev. 2014, 33, 511–525. [Google Scholar] [CrossRef] [Green Version]
- D’Amico, L.; Roato, I. The Impact of Immune System in Regulating Bone Metastasis Formation by Osteotropic Tumors. J. Immunol. Res. 2015, 2015, 143526. [Google Scholar] [CrossRef]
- Kawai, T.; Matsuyama, T.; Hosokawa, Y.; Makihira, S.; Seki, M.; Karimbux, N.Y.; Goncalves, R.B.; Valverde, P.; Dibart, S.; Li, Y.-P.; et al. B and T Lymphocytes Are the Primary Sources of RANKL in the Bone Resorptive Lesion of Periodontal Disease. Am. J. Pathol. 2006, 169, 987–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Qin, L.; Bergenstock, M.; Bevelock, L.M.; Novack, D.V.; Partridge, N.C. Parathyroid Hormone Stimulates Osteoblastic Expression of MCP-1 to Recruit and Increase the Fusion of Pre/Osteoclasts. J. Biol. Chem. 2007, 282, 33098–33106. [Google Scholar] [CrossRef] [Green Version]
- Titanji, K.; Vunnava, A.; Sheth, A.N.; Delille, C.; Lennox, J.L.; Sanford, S.E.; Foster, A.; Knezevic, A.; Easley, K.; Weitzmann, M.N.; et al. Dysregulated B Cell Expression of RANKL and OPG Correlates with Loss of Bone Mineral Density in HIV Infection. PLoS Pathog. 2014, 10, e1004497. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Rolle, S.; Wellner, M.; Cardoso, M.C.; Scheidereit, C.; Luft, F.C.; Kettritz, R. Inhibition of NF-κB by a TAT-NEMO–binding domain peptide accelerates constitutive apoptosis and abrogates LPS-delayed neutrophil apoptosis. Blood 2003, 102, 2259–2267. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Suematsu, A.; Okamoto, K.; Yamaguchi, A.; Morishita, Y.; Kadono, Y.; Tanaka, S.; Kodama, T.; Akira, S.; Iwakura, Y.; et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J. Exp. Med. 2006, 203, 2673–2682. [Google Scholar] [CrossRef] [Green Version]
- Toraldo, G.; Roggia, C.; Qian, W.-P.; Pacifici, R.; Weitzmann, M.N. IL-7 induces bone loss in vivo by induction of receptor activator of nuclear factor κB ligand and tumor necrosis factor α from T cells. Proc. Natl. Acad. Sci. USA 2002, 100, 125–130. [Google Scholar] [CrossRef] [Green Version]
- Cenci, S.; Weitzmann, M.N.; Roggia, C.; Namba, N.; Novack, D.; Woodring, J.; Pacifici, R. Estrogen deficiency induces bone loss by enhancing T-cell production of TNF-α. J. Clin. Investig. 2000, 106, 1229–1237. [Google Scholar] [CrossRef] [Green Version]
- Buchwald, Z.S.; Kiesel, J.R.; Yang, C.; DiPaolo, R.; Novack, D.V.; Aurora, R. Osteoclast-induced Foxp3+ CD8 T-cells limit bone loss in mice. Bone 2013, 56, 163–173. [Google Scholar] [CrossRef] [Green Version]
- Chang, M.K.; Raggatt, L.-J.; Alexander, K.A.; Kuliwaba, J.S.; Fazzalari, N.L.; Schroder, K.; Maylin, E.R.; Ripoll, V.M.; Hume, D.A.; Pettit, A.R. Osteal Tissue Macrophages Are Intercalated throughout Human and Mouse Bone Lining Tissues and Regulate Osteoblast Function In Vitro and In Vivo. J. Immunol. 2008, 181, 1232–1244. [Google Scholar] [CrossRef] [Green Version]
- Morris, E.V.; Edwards, C.M. Bone Marrow Adipose Tissue: A New Player in Cancer Metastasis to Bone. Front. Endocrinol. 2016, 7, 90. [Google Scholar] [CrossRef]
- Caers, J.; Deleu, S.; Belaid, Z.; De Raeve, H.; Van Valckenborgh, E.; De Bruyne, E.; Defresne, M.-P.; Van Riet, I.; Van Camp, B.; Vanderkerken, K. Neighboring adipocytes participate in the bone marrow microenvironment of multiple myeloma cells. Leukemia 2007, 21, 1580–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, J.; Onal, M.; Jilka, R.L.; Weinstein, R.S.; Manolagas, S.C.; O’Brien, C.A. Matrix-embedded cells control osteoclast formation. Nat. Med. 2011, 17, 1235–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weilbaecher, K.N.; Guise, T.A.; McCauley, L.K. Cancer to bone: A fatal attraction. Nat. Rev. Cancer 2011, 11, 411–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ell, B.; Mercatali, L.; Ibrahim, T.; Campbell, N.; Schwarzenbach, H.; Pantel, K.; Amadori, D.; Kang, Y. Tumor-Induced Osteoclast miRNA Changes as Regulators and Biomarkers of Osteolytic Bone Metastasis. Cancer Cell 2013, 24, 542–556. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordón-Cardo, C.; Guise, T.A.; Massagué, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Martin, T.J. Parathyroid Hormone-Related Protein, Its Regulation of Cartilage and Bone Development, and Role in Treating Bone Diseases. Physiol. Rev. 2016, 96, 831–871. [Google Scholar] [CrossRef] [Green Version]
- Andrade, K.; Fornetti, J.; Zhao, L.; Miller, S.C.; Randall, R.L.; Anderson, N.; Waltz, S.E.; McHale, M.; Welm, A.L. RON kinase: A target for treatment of cancer-induced bone destruction and osteoporosis. Sci. Transl. Med. 2017, 9, eaai9338. [Google Scholar] [CrossRef] [Green Version]
- A Guise, T.; Yin, J.J.; Taylor, S.D.; Kumagai, Y.; Dallas, M.; Boyce, B.F.; Yoneda, T.; Mundy, G.R. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J. Clin. Investig. 1996, 98, 1544–1549. [Google Scholar] [CrossRef]
- Roodman, G.D. Biology of Osteoclast Activation in Cancer. J. Clin. Oncol. 2001, 19, 3562–3571. [Google Scholar] [CrossRef]
- Fornetti, J.; Welm, A.L.; A Stewart, S. Understanding the Bone in Cancer Metastasis. J. Bone Miner. Res. 2018, 33, 2099–2113. [Google Scholar] [CrossRef]
- Mohammad, K.S.; Guise, T.A. Mechanisms of Osteoblastic Metastases: Role of Endothelin-1. Clin. Orthop. Relat. Res. 2003, 415, S67–S74. [Google Scholar] [CrossRef] [PubMed]
- Reddi, A. Bone and cartilage differentiation. Curr. Opin. Genet. Dev. 1994, 4, 737–744. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, T.L.; Ji, C.; Chen, Y.; Kim, K.K.; Imagawa, M.; Ito, Y.; Centrella, M. Runt Domain Factor (Runx)-dependent Effects on CCAAT/Enhancer-binding Protein δ Expression and Activity in Osteoblasts. J. Biol. Chem. 2000, 275, 21746–21753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, J.J.; Pollock, C.B.; Kelly, K. Mechanisms of cancer metastasis to the bone. Cell Res. 2005, 15, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, H.; Feng, C.-C.; Ding, G.-X.; Meng, D.-L.; Ding, Q.; Fang, Z.-J.; Xia, G.-W.; Xu, G.; Jiang, H.-W. Med19 promotes bone metastasis and invasiveness of bladder urothelial carcinoma via bone morphogenetic protein 2. Ann. Diagn. Pathol. 2013, 17, 259–264. [Google Scholar] [CrossRef]
- Wu, K.; Fan, J.; Zhang, L.; Ning, Z.; Zeng, J.; Zhou, J.; Li, L.; Chen, Y.; Zhang, T.; Wang, X.; et al. PI3K/Akt to GSK3β/β-catenin signaling cascade coordinates cell colonization for bladder cancer bone metastasis through regulating ZEB1 transcription. Cell. Signal. 2012, 24, 2273–2282. [Google Scholar] [CrossRef]
- Yang, Z.-J.; Liu, F.-X.; Yang, Y.-S.; Yang, X.; Zhu, G.-X. Expression of bone-morphogenetic protein 2 and tumor necrosis factor α correlates with bone metastases in bladder urothelial carcinoma. Ann. Diagn. Pathol. 2012, 17, 51–53. [Google Scholar] [CrossRef]
- Chan, E.; Patel, A.; Heston, W.; Larchian, W. Mouse orthotopic models for bladder cancer research. Br. J. Urol. 2009, 104, 1286–1291. [Google Scholar] [CrossRef]
- Chikazawa, M.; Inoue, K.; Fukata, S.; Karashima, T.; Shuin, T. Expression of Angiogenesis-Related Genes Regulates Different Steps in the Process of Tumor Growth and Metastasis in Human Urothelial Cell Carcinoma of the Urinary Bladder. Pathobiology 2008, 75, 335–345. [Google Scholar] [CrossRef]
- Nitz, M.D.; Harding, M.A.; Theodorescu, D. Invasion and Metastasis Models for Studying RhoGDI2 in Bladder Cancer. Methods Enzymol. 2008, 439, 219–233. [Google Scholar] [CrossRef]
- Sato, K.; Yuasa, T.; Nogawa, M.; Kimura, S.; Segawa, H.; Yokota, A.; Maekawa, T. A third-generation bisphosphonate, minodronic acid (YM529), successfully prevented the growth of bladder cancer in vitro and in vivo. Br. J. Cancer 2006, 95, 1354–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mundy, G. Preclinical models of bone metastases. Semin. Oncol. 2001, 28, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.R.; Chan, E.S.; Hansel, D.E.; Powell, C.T.; Heston, W.D.; Larchian, W.A. Transabdominal Micro-ultrasound Imaging of Bladder Cancer in a Mouse Model: A Validation Study. Urology 2010, 75, 799–804. [Google Scholar] [CrossRef]
- Kaijzel, E.L.; van der Pluijm, G.; Löwik, C.W. Whole-Body Optical Imaging in Animal Models to Assess Cancer Development and Progression. Clin. Cancer Res. 2007, 13, 3490–3497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriquez, N.V.; Van Overveld, P.G.M.; Que, I.; Buijs, J.T.; Bachelier, R.; Kaijzel, E.L.; Löwik, C.W.G.M.; Clézardin, P.; Van Der Pluijm, G. Advances in optical imaging and novel model systems for cancer metastasis research. Clin. Exp. Metast. 2007, 24, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Black, P.C.; Shetty, A.; Brown, G.A.; Metwalli, A.R.; Agarwal, P.K.; McConkey, D.J.; Hazle, J.D.; Dinney, C.P.; Esparza-Coss, E. Validating bladder cancer xenograft bioluminescence with magnetic resonance imaging: The significance of hypoxia and necrosis. Br. J. Urol. 2010, 106, 1799–1804. [Google Scholar] [CrossRef]
- Hadaschik, B.A.; Black, P.C.; Sea, J.C.; Metwalli, A.R.; Fazli, L.; Dinney, C.P.; Gleave, M.E.; So, A.I. A validated mouse model for orthotopic bladder cancer using transurethral tumour inoculation and bioluminescence imaging. Br. J. Urol. 2007, 100, 1377–1384. [Google Scholar] [CrossRef]
- Nogawa, M.; Yuasa, T.; Kimura, S.; Tanaka, M.; Kuroda, J.; Sato, K.; Yokota, A.; Segawa, H.; Toda, Y.; Kageyama, S.; et al. Intravesical administration of small interfering RNA targeting PLK-1 successfully prevents the growth of bladder cancer. J. Clin. Investig. 2005, 115, 978–985. [Google Scholar] [CrossRef] [Green Version]
- Chong, L.; Ruping, Y.; Jiancheng, B.; Guohong, Y.; Yougang, F.; Jiansong, W.; Xiang, G.; Jie, H.; Shusheng, X. Characterization of a novel transplantable orthotopic murine xenograft model of a human bladder transitional cell tumor (BIU-87). Cancer Biol. Ther. 2006, 5, 394–398. [Google Scholar] [CrossRef] [Green Version]
- Van der Horst, G.; van Asten, J.J.; Figdor, A.; Hoogen, C.V.D.; Cheung, H.; Bevers, R.F.; Pelger, R.; van der Pluijm, G. Real-Time Cancer Cell Tracking by Bioluminescence in a Preclinical Model of Human Bladder Cancer Growth and Metastasis. Eur. Urol. 2011, 60, 337–343. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Dow, A.C.; Kupiec-Weglinski, J.; Busuttil, R.W.; Lipshutz, G.S. Evaluation of Gene Promoters for Liver Expression by Hydrodynamic Gene Transfer. J. Surg. Res. 2008, 148, 60–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheelock, M.C.; O’Conor, V.J. Metastasis in tibia from carcinoma of the urinary bladder. Q. Bull. Northwestern Univ. (Evanston Ill.) Med. Sch. 1953, 27, 111–113. [Google Scholar]
- Hong, J.H. Early isolated bone metastases without local recurrence in non-muscle invasive bladder cancer. Int. J. Surg. Case Rep. 2015, 10, 41–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, Y.; Oi, H.; Oyama, T.; Kagawa, J.; Komori, M.; Senzaki, T.; Fukawa, T.; Takahashi, H.; Takemura, M.; Yamaguchi, K.; et al. Non-muscle invasive bladder cancer with multiple bone metastasis without local invasion: A case report. Hinyokika Kiyo Acta Urol. Jpn. 2013, 59, 669–672. [Google Scholar]
- von Moos, R.; Costa, L.; Gonzalez-Suarez, E.; Terpos, E.; Niepel, D.; Body, J. Management of bone health in solid tumours: From bisphosphonates to a monoclonal antibody. Cancer Treat. Rev. 2019, 76, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Coleman, R.E. Clinical Features of Metastatic Bone Disease and Risk of Skeletal Morbidity. Clin. Cancer Res. 2006, 12, 6243s–6249s. [Google Scholar] [CrossRef] [Green Version]
- Clines, G.A.; Guise, T.A. Molecular mechanisms and treatment of bone metastasis. Expert Rev. Mol. Med. 2008, 10, e7. [Google Scholar] [CrossRef]
- Coleman, R.E.; Croucher, P.I.; Padhani, A.R.; Clézardin, P.; Chow, E.; Fallon, M.; Guise, T.; Colangeli, S.; Capanna, R.; Costa, L. Bone metastases. Nat. Rev. Dis. Prim. 2020, 6, 1–28. [Google Scholar] [CrossRef]
- Jarvik, J.G.; Deyo, R.A. Diagnostic Evaluation of Low Back Pain with Emphasis on Imaging. Ann. Intern. Med. 2002, 137, 586–597. [Google Scholar] [CrossRef]
- Togral, G.; Arikan, M.; Aktas, E.; Öztürk, R.; Guven, O.; Eksioglu, F. Surgical management of bone metastases from urological malignancies: Analyses of 70 cases. Acta Orthop. Traumatol. Turc. 2015, 49, 634–640. [Google Scholar] [CrossRef]
- Thomson, V.; Pialat, J.-B.; Gay, F.; Coulon, A.; Voloch, A.; Granier, A.; Guérin, J.-C.; Viallon, M.; Berthezene, Y. Whole-Body MRI for Metastases Screening: A Preliminary Study Using 3D VIBE Sequences with Automatic Subtraction between Noncontrast and Contrast Enhanced Images. Am. J. Clin. Oncol. 2008, 31, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.; Hadji, P.; Body, J.-J.; Santini, D.; Chow, E.; Terpos, E.; Oudard, S.; Bruland, P.; Flamen, P.; Kurth, A.; et al. Bone health in cancer: ESMO Clinical Practice Guidelines. Ann. Oncol. 2020, 31, 1650–1663. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.; Demers, L.M.; Gouveia-Oliveira, A.; Schaller, J.; Costa, E.B.; De Moura, M.C.; Lipton, A. Prospective Evaluation of the Peptide-Bound Collagen Type I Cross-Links N-Telopeptide and C-Telopeptide in Predicting Bone Metastases Status. J. Clin. Oncol. 2002, 20, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Glas, A.S.; Roos, D.; Deutekom, M.; Zwinderman, A.H.; Bossuyt, P.M.; Kurth, K.H. Tumor Markers in the Diagnosis of Primary Bladder Cancer. A Systematic Review. J. Urol. 2003, 169, 1975–1982. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, X.; Su, L.-J.; Flaig, T.W. Pazopanib Synergizes with Docetaxel in the Treatment of Bladder Cancer Cells. Urology 2011, 78, 233.e7–233.e13. [Google Scholar] [CrossRef] [Green Version]
- Nadal, R.; Bellmunt, J. Management of metastatic bladder cancer. Cancer Treat. Rev. 2019, 76, 10–21. [Google Scholar] [CrossRef]
- Xiong, W.; Qi, L.; Jiang, N.; Zhao, Q.; Chen, L.; Jiang, X.; Li, Y.; Zhou, Z.; Shen, J. Metformin Liposome-Mediated PD-L1 Downregulation for Amplifying the Photodynamic Immunotherapy Efficacy. ACS Appl. Mater. Interfaces 2021, 13, 8026–8041. [Google Scholar] [CrossRef]
- Donin, N.M.; Lenis, A.T.; Holden, S.; Drakaki, A.; Pantuck, A.; Belldegrun, A.; Chamie, K. Immunotherapy for the Treatment of Urothelial Carcinoma. J. Urol. 2016, 197, 14–22. [Google Scholar] [CrossRef]
- Bednova, O.; Leyton, J.V. Targeted Molecular Therapeutics for Bladder Cancer—A New Option beyond the Mixed Fortunes of Immune Checkpoint Inhibitors? Int. J. Mol. Sci. 2020, 21, 7268. [Google Scholar] [CrossRef]
- Robinson, B.D.; Vlachostergios, P.J.; Bhinder, B.; Liu, W.; Li, K.; Moss, T.J.; Bareja, R.; Park, K.; Tavassoli, P.; Cyrta, J.; et al. Upper tract urothelial carcinoma has a luminal-papillary T-cell depleted contexture and activated FGFR3 signaling. Nat. Commun. 2019, 10, 2977. [Google Scholar] [CrossRef] [Green Version]
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2019, 381, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Sarfaty, M.; Rosenberg, J.E. Antibody-Drug Conjugates in Urothelial Carcinomas. Curr. Oncol. Rep. 2020, 22, 13. [Google Scholar] [CrossRef] [PubMed]
- Ebetino, F.H.; Hogan, A.-M.L.; Sun, S.; Tsoumpra, M.K.; Duan, X.; Triffitt, J.T.; Kwaasi, A.A.; Dunford, J.E.; Barnett, B.L.; Oppermann, U.; et al. The relationship between the chemistry and biological activity of the bisphosphonates. Bone 2011, 49, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Roelofs, A.J.; Thompson, K.; Gordon, S.; Rogers, M.J. Molecular Mechanisms of Action of Bisphosphonates: Current Status. Clin. Cancer Res. 2006, 12, 6222s–6230s. [Google Scholar] [CrossRef] [Green Version]
- Body, J.-J.; Diel, I.J.; Lichinitzer, M.; Lazarev, A.; Pecherstorfer, M.; Bell, R.H.; Tripathy, D.; Bergström, B. Oral ibandronate reduces the risk of skeletal complications in breast cancer patients with metastatic bone disease: Results from two randomised, placebo-controlled phase III studies. Br. J. Cancer 2004, 90, 1133–1137. [Google Scholar] [CrossRef]
- Rosen, L.S.; Gordon, D.; Tchekmedyian, N.S.; Yanagihara, R.; Hirsh, V.; Krzakowski, M.; Pawlicki, M.; de Souza, P.; Zheng, M.; Urbanowitz, G.; et al. Long-term efficacy and safety of zoledronic acid in the treatment of skeletal metastases in patients with nonsmall cell lung carcinoma and other solid tumors. Cancer 2004, 100, 2613–2621. [Google Scholar] [CrossRef]
- Zaghloul, M.S.; Boutrus, R.; El-Hossieny, H.; Kader, Y.A.; El-Attar, I.; Nazmy, M. A prospective, randomized, placebo-controlled trial of zoledronic acid in bony metastatic bladder cancer. Int. J. Clin. Oncol. 2010, 15, 382–389. [Google Scholar] [CrossRef]
- Lacey, D.L.; Boyle, W.J.; Simonet, W.S.; Kostenuik, P.J.; Dougall, W.C.; Sullivan, J.K.; Martin, J.S.; Dansey, R. Bench to bedside: Elucidation of the OPG–RANK–RANKL pathway and the development of denosumab. Nat. Rev. Drug Discov. 2012, 11, 401–419. [Google Scholar] [CrossRef]
- Kostenuik, P.J.; Nguyen, H.Q.; McCabe, J.; Warmington, K.S.; Kurahara, C.; Sun, N.; Chen, C.; Li, L.; Cattley, R.C.; Van, G.; et al. Denosumab, a Fully Human Monoclonal Antibody to RANKL, Inhibits Bone Resorption and Increases BMD in Knock-In Mice That Express Chimeric (Murine/Human) RANKL*. J. Bone Miner. Res. 2009, 24, 182–195. [Google Scholar] [CrossRef]
- Giuliani, N.; Colla, S.; Sala, R.; Moroni, M.; Lazzaretti, M.; La Monica, S.; Bonomini, S.; Hojden, M.; Sammarelli, G.; Barille-Nion, S.; et al. Human myeloma cells stimulate the receptor activator of nuclear factor-κB ligand (RANKL) in T lymphocytes: A potential role in multiple myeloma bone disease. Blood 2002, 100, 4615–4621. [Google Scholar] [CrossRef] [Green Version]
- Luger, N.M.; Honore, P.; A Sabino, M.; Schwei, M.J.; Rogers, S.D.; Mach, D.B.; Clohisy, D.R.; Mantyh, P.W. Osteoprotegerin diminishes advanced bone cancer pain. Cancer Res. 2001, 61, 4038–4047. [Google Scholar] [PubMed]
- Roudier, M.P.; Bain, S.D.; Dougall, W.C. Effects of the RANKL inhibitor, osteoprotegerin, on the pain and histopathology of bone cancer in rats. Clin. Exp. Metast. 2006, 23, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Oyajobi, B.O.; Anderson, D.M.; Traianedes, K.; Williams, P.J.; Yoneda, T.; Mundy, G.R. Therapeutic efficacy of a soluble receptor activator of nuclear factor kappaB-IgG Fc fusion protein in suppressing bone resorption and hypercalcemia in a model of humoral hypercalcemia of malignancy. Cancer Res. 2001, 61, 2572–2578. [Google Scholar] [PubMed]
- Fizazi, K.; Carducci, M.; Smith, M.; Damião, R.; Brown, J.; Karsh, L.; Milecki, P.; Shore, N.; Rader, M.; Wang, H.; et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: A randomised, double-blind study. Lancet 2011, 377, 813–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, D.H.; Costa, L.; Goldwasser, F.; Hirsh, V.; Hungria, V.; Prausova, J.; Scagliotti, G.V.; Sleeboom, H.; Spencer, A.; Vadhan-Raj, S.; et al. Randomized, Double-Blind Study of Denosumab Versus Zoledronic Acid in the Treatment of Bone Metastases in Patients with Advanced Cancer (Excluding Breast and Prostate Cancer) or Multiple Myeloma. J. Clin. Oncol. 2011, 29, 1125–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Sartor, O.; Hoskin, P.; Bruland, Ø.S. Targeted radio-nuclide therapy of skeletal metastases. Cancer Treat. Rev. 2013, 39, 18–26. [Google Scholar] [CrossRef]
- Tsourdi, E.; Langdahl, B.; Cohen-Solal, M.; Aubry-Rozier, B.; Eriksen, E.F.; Guañabens, N.; Obermayer-Pietsch, B.; Ralston, S.H.; Eastell, R.; Zillikens, M.C. Discontinuation of Denosumab therapy for osteoporosis: A systematic review and position statement by ECTS. Bone 2017, 105, 11–17. [Google Scholar] [CrossRef]
- Lamy, O.; Stoll, D.; Aubry-Rozier, B.; Rodriguez, E.G. Stopping Denosumab. Curr. Osteoporos. Rep. 2019, 17, 8–15. [Google Scholar] [CrossRef]
- Parker, C.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fosså, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M.; et al. Alpha Emitter Radium-223 and Survival in Metastatic Prostate Cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, S.; Franzén, L.; Parker, C.; Tyrrell, C.; Blom, R.; Tennvall, J.; Lennernäs, B.; Petersson, U.; Johannessen, D.C.; Sokal, M.; et al. Bone-targeted radium-223 in symptomatic, hormone-refractory prostate cancer: A randomised, multicentre, placebo-controlled phase II study. Lancet Oncol. 2007, 8, 587–594. [Google Scholar] [CrossRef] [PubMed]
- McDonald, R.; Ding, K.; Brundage, M.; Meyer, R.M.; Nabid, A.; Chabot, P.; Coulombe, G.; Ahmed, S.; Kuk, J.; Dar, A.R.; et al. Effect of Radiotherapy on Painful Bone Metastases. JAMA Oncol. 2017, 3, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Hird, A.; Chow, E.; Zhang, L.; Wong, R.; Wu, J.; Sinclair, E.; Danjoux, C.; Tsao, M.; Barnes, E.; Loblaw, A. Determining the Incidence of Pain Flare Following Palliative Radiotherapy for Symptomatic Bone Metastases: Results from Three Canadian Cancer Centers. Int. J. Radiat. Oncol. 2009, 75, 193–197. [Google Scholar] [CrossRef]
- Chow, E.; Meyer, R.M.; Ding, K.; Nabid, A.; Chabot, P.; Wong, P.; Ahmed, S.; Kuk, J.; Dar, A.R.; Mahmud, A.; et al. Dexamethasone in the prophylaxis of radiation-induced pain flare after palliative radiotherapy for bone metastases: A double-blind, randomised placebo-controlled, phase 3 trial. Lancet Oncol. 2015, 16, 1463–1472. [Google Scholar] [CrossRef] [PubMed]
- Sprave, T.; Verma, V.; Förster, R.; Schlampp, I.; Bruckner, T.; Bostel, T.; Welte, S.E.; Tonndorf-Martini, E.; Nicolay, N.H.; Debus, J.; et al. Randomized phase II trial evaluating pain response in patients with spinal metastases following stereotactic body radiotherapy versus three-dimensional conformal radiotherapy. Radiother. Oncol. 2018, 128, 274–282. [Google Scholar] [CrossRef] [Green Version]
- Sopata, M.; Katz, N.; Carey, W.; Smith, M.D.; Keller, D.; Verburg, K.M.; West, C.R.; Wolfram, G.; Brown, M.T. Efficacy and safety of tanezumab in the treatment of pain from bone metastases. Pain 2015, 156, 1703–1713. [Google Scholar] [CrossRef]
- Arendt-Nielsen, L.; Harris, S.; Whiteside, G.T.; Hummel, M.; Knappenberger, T.; O’Keefe, S.; Kapil, R.; Kyle, D. A randomized, double-blind, positive-controlled, 3-way cross-over human experimental pain study of a TRPV1 antagonist (V116517) in healthy volunteers and comparison with preclinical profile. Pain 2016, 157, 2057–2067. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yi, L.; Ai, K.; Li, X.; Li, Z.; Li, Y. Bone Metastasis in Bladder Cancer. J. Pers. Med. 2023, 13, 54. https://doi.org/10.3390/jpm13010054
Yi L, Ai K, Li X, Li Z, Li Y. Bone Metastasis in Bladder Cancer. Journal of Personalized Medicine. 2023; 13(1):54. https://doi.org/10.3390/jpm13010054
Chicago/Turabian StyleYi, Lei, Kai Ai, Xurui Li, Zhihong Li, and Yuan Li. 2023. "Bone Metastasis in Bladder Cancer" Journal of Personalized Medicine 13, no. 1: 54. https://doi.org/10.3390/jpm13010054