1. Introduction
The A-to-G transition at nucleotide 8344 (m.8344A > G) of mtDNA is the prevalent mutation found in a multisystem disorder, and is known with the acronym MERRF (myoclonus epilepsy with ragged-red fibers). It is characterized by myoclonus, generalized epilepsy, ataxia, weakness, dementia as well as signs of multisystem involvement [
1,
2,
3]. The histopathological study of the skeletal muscle tissue typically shows ragged-red fibers (RRFs) with the modified Gomori trichrome (MGT) stain and hyperactive fibers with the succinate dehydrogenase (SDH) stain. Histochemical reaction for cytochrome c oxidase (COX) shows lack of activity in RRFs and some non-RRFs [
4,
5,
6]. Occasionally, RRFs may not be observed [
7]. The presence of lipomas has often been reported in patients affected with MERRF and/or in their maternally-related family members [
8,
9,
10].
Moreover, the m.8344A > G variant has been reported in association with isolated myopathy, lipomatosis with muscle lipid storage, or Leigh syndrome [
11,
12,
13]. Other unusual manifestations include sudden infant death syndrome [
14], spasmodic dysphonia [
15], Parkinsonism with neuropathy and myopathy [
16], infantile-onset ataxia, myoclonus and bilateral putaminal necrosis on brain MRI [
17], sudden respiratory failure in adulthood [
18], acute central and peripheral nervous system demyelinating disease [
19], and typical MELAS (mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes) picture [
20], reflecting the heterogenous clinical presentations associated resulting from mtDNA defects.
The pathologic mutation affects all tissues; however, since mtDNA mutations are heteroplasmic, the variable tissue distribution of mutated mtDNA usually occurs in the same individual. The m.8344A > G variant is usually heteroplasmic in tissues collected from classical MERRF patients and the level of mutational load required to display a biochemical phenotype (biochemical threshold) is in the range between 60 and 90%, suggesting a moderate detrimental behavior for this nucleotide change. Both the heteroplasmy and the selective tissue vulnerability to impaired oxidative metabolism (skeletal/cardiac muscle and brain have a higher energetic demand) are important factors in determining the clinical expression of mtDNA mutations. These aspects, along with the different regional levels of mutant DNA, the compensatory increase in global mtDNA content, and the presence of nuclear modifiers, hamper the establishment of a clear correlation between the genotype and clinical phenotype [
21,
22].
Although there may be a high clinical variability among members of the same family, central nervous system (CNS) manifestations have always been reported throughout family generations. In this study, we describe a patient carrying the m.8344A > G mutation in mitochondrial DNA, presenting with late onset myopathy, multiple lipomas, and diabetes.
Neither the proband nor the other affected family members show signs of CNS involvement.
2. Case Report
A 53-year-old man approached our Neuromuscular Unit due to an incidental finding of hyperckemia (between 300 and 400 U/L, n.v. < 180 U/L). He was diabetic and had a few lipomas, but otherwise asymptomatic. Almost 2 years later, he developed fatigue which progressively worsened. CK levels had moderately increased to 970 U/L. The neurological examination was normal and his past medical history was unremarkable.
The patient’s mother, who had died at 77 years of age, was diabetic, cardiopathic, and displayed multiple lipomas. Proband’s sisters, aged 60 and 50 years, presented normal serum CK levels. The elder sister has mild bilateral eyelid ptosis and had suffered from thyroiditis; while the younger sister had undergone surgery for colon cancer at the age of 48 years and the follow-up was negative.
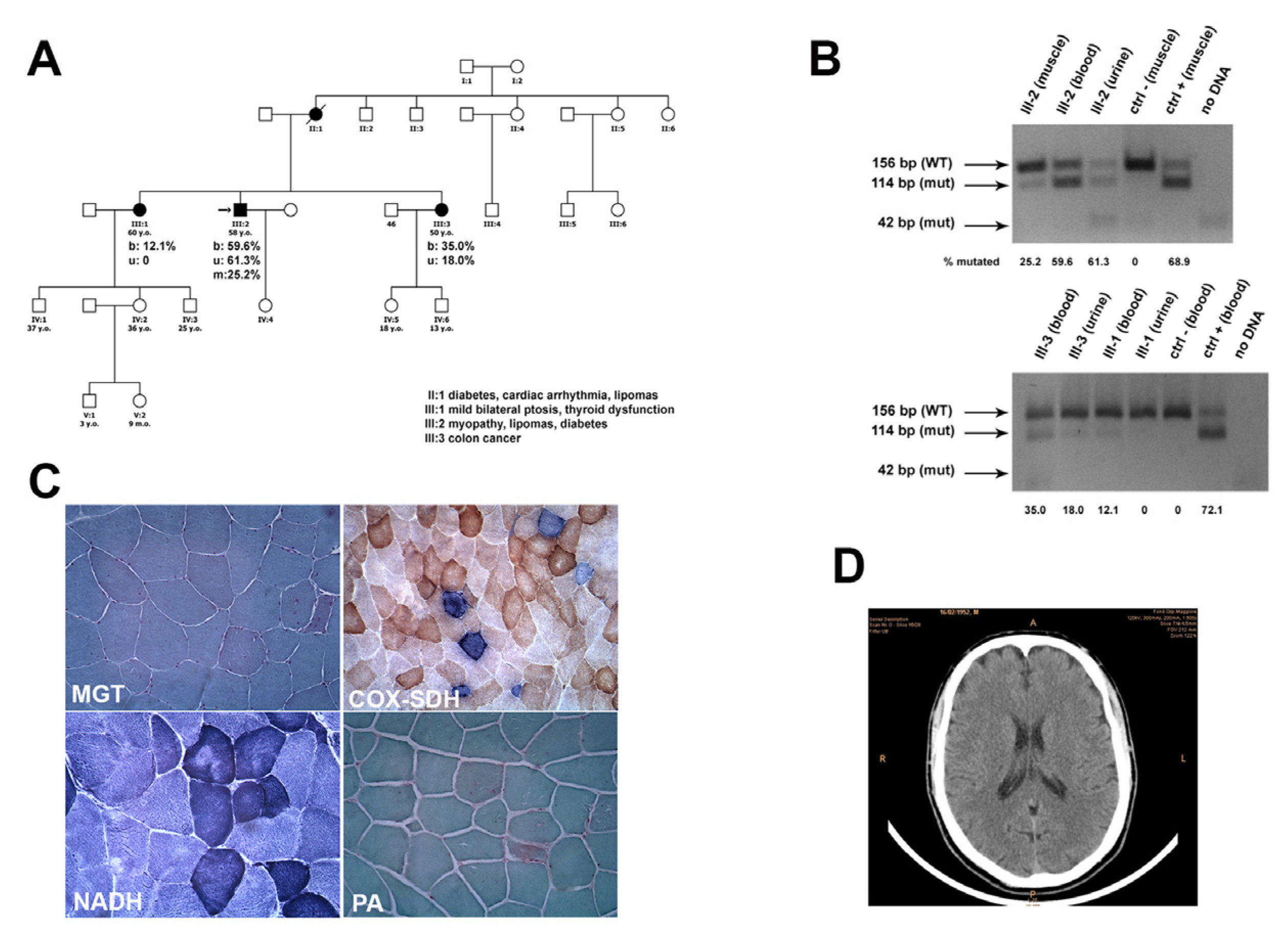
The first sister has three children, two males and one female, aged 33, 23, and 32 years, respectively. The 33-year-old son has two children, one male and one female. The 32-year-old daughter has a son. The second sister has a 13-year-old daughter and a son aged 11 years. They are all reported asymptomatic (
Figure 1A). Descendants of two maternal aunts are reported healthy and did not undergo any genetic evaluation.
After an EMG examination, which showed a myopathic pattern in proximal four-limb muscles, the proband underwent left biceps skeletal muscle biopsy. Following plain brain CT scan (
Figure 1D) and EEG, both examinations were normal.
Moreover, we evaluated both sisters whose neurological examination was normal except for the mild eyelid ptosis in the elder one. Blood and urinary samples were taken from both subjects for DNA extraction.
3. Materials and Methods
After the patient had signed a written informed consent on 28 February 2005, a skeletal muscle specimen (biopsy code number: 96974) from his left biceps brachii muscle was obtained by an open biopsy, according to a protocol approved by the Institutional Review Board of the “IRCCS Ca’ Granda Foundation Ospedale Maggiore Policlinico, Italy”.
A total of 8 µm-thick cryostatic sections were processed according to standard histological and histochemical techniques [
23]. Mitochondrial enzymatic activity was demonstrated by COX, SDH, and double COX-SDH staining [
23]. Enzymatic evaluations for acid phosphatase (AP), phosphofructokinase (PFK), myophosphorylase (PYGM), and myoadenylate deaminase (MAD) were also performed.
Immunohistochemistry for sarcolemmal proteins (dystrophin, alpha-, and gamma-sarcoglycan) and for an evaluation of possible inflammatory signs were performed [
24].
Furthermore, after obtaining written consent, genomic DNA was extracted from peripheral blood, urine, and muscle from both the proband (muscle, blood, urine) and his sisters (blood, urine). The extracted mtDNA was PCR-amplified using MitoSEQ Resequencing System (Applied Biosystem, FosterCity, CA, USA) and sequenced on an ABI PRISM 3100 Genetic Analyzer (Applied Biosystem).
Mutational loads in the patient’s tissues were assessed by PCR-RFLP performed using a modified primer that creates a
BglI-restriction site in mutant molecules. Aliquots of PCR products were digested and electrophoresed on a 4% agarose gel. The proportion of mutant mtDNA was evaluated by densitometry using the NIH ImageJ 2.1.0 software (NIH National Institute of Health, Bethesda, MD, USA) (
https://imagej.nih.gov/ij/download.html, accessed on 5 November 2022) in the month of September 2011.
4. Results
Sections stained with MGT showed quite a few ragged-red fibers. Histochemical reactions for mitochondrial enzymatic activity showed several COX-negative fibers, many of which were intensely stained with SDH (RRFs) (
Figure 1C). Glycogen content was normal and no lipid storage was present.
Enzymatic activities for PKF, PYGM, and MAD were normal. The staining for PA showed a slightly increased subsarcolemmal signal in a few fibers. Immunohistochemistry for sarcolemmal proteins was normal (data not shown).
Genetic analysis of the entire mtDNA sequence in proband’s muscle revealed the presence of the A8344G mtDNA mutation. The same mutation was detected in the two sisters.
The degree of heteroplasmy of this mutation was analyzed in proband’s skeletal muscle and in blood leukocytes and urinary sediment samples from both the proband and his sisters. Densitometric analysis in the patient’s tissues revealed that the A8344G mutation accounted for 25.2% of the total mtDNA in muscle, 60% in blood, and 61% in urine (
Figure 1B). The younger sister presented a mutational load of 35% in blood and 18% in urine, while the elder sister had 12.1% mtDNA mutation in blood, but no mutation was detected in urine. Detailed quantification of the mutational load in the family members is presented in
Figure 1B.
5. Discussion
MERRF syndrome is a devastating neuromuscular disorder characterized by myoclonic epilepsy, generalized weakness, muscle wasting, cerebellar ataxia, deafness, dementia, and RRFs at muscle biopsy. It is transmitted through maternal lineage [
1,
2,
5]. About 80% of MERRF cases are caused by the A8344G mutation in the tRNA Lys gene, [
3,
25] although a few less frequent mtDNA point mutations have also been found in MERRF patients [
26,
27,
28,
29,
30].
Our patient did present RRFs and lipomas, the latter was also diagnosed in his mother. These findings, along with the presence of other features indicating multisystem involvement in both patient and family (diabetes, cardiopathy, endocrine dysfunction) as well as lack of lipid storage at muscle biopsy, prompted a diagnosis of atypical MERRF syndrome.
Our patient showed an atypical clinical presentation, with isolated hyperckemia at onset followed by development of myopathy in his fifties. The associated presence of maternally inherited diabetes and lipomas suggested a diagnosis of mitochondrial disorder, which was confirmed by both morphological and biomolecular findings.
Lipomas have often been reported in patients bearing the A8344G mutation in association with MERRF syndrome or other central nervous system involvement [
8,
9,
10,
31,
32]. Indeed, the presence of maternally inherited lipomas associated with the involvement of other organs/systems is an almost unequivocal indication of the presence of mutations in the tRNA Lys. It is not known how an impaired mitochondrial function, due to mutations in tRNA Lys, causes this effect on adipose tissue; however, there is evidence that mitochondrial function is important for normal development of adipose tissue in humans [
33].
The A8344G mutation has been considered a relatively “benign” mutation, since a high degree of mutational load is required to produce clinical manifestations. In skeletal muscle tissue, the threshold level beyond which the pathological phenotype becomes evident is estimated to be higher than 60% mutational load [
34]. Indeed, it has been suggested that approximately 15% of residual wildtype mtDNA is sufficient to restore translation and COX activity to near-normal levels, thus “rescuing” the clinical phenotype. Interestingly, our patient had a low mutational load in muscle (25%), and this can explain the late onset of symptoms.
Furthermore, the A8344G mutation is usually present in high proportion in DNA from urine and blood, the mutational load being ordinarily higher in urine than in blood [
35]. In accordance with the data reported in the literature, our patient has a mutational load of 61% in urine and 60% in blood; however, both his sisters have a higher mutant load in blood than in urine. We were unable to establish the mutational load in the lipomas since the patient refused to undergo lipoma biopsy.
In our family, we established a certain positive correlation between the severity of clinical signs and instrumental evidence. Indeed, only the proband, who, unlike his sisters, has increased serum CK levels, is symptomatic. We could not make any correlations in terms of skeletal muscle mutational load since the two sisters did not undergo skeletal muscle biopsy [
31,
32,
33].
Muscle biopsy showed typical histopathological features of MERRF [
5,
6,
36], in particular the presence of RRFs at MGT, confirmed by increased SDH activity, the absence of COX activity in most RRFs, and a number of COX-negative/deficient non-RRFs.
The absence of central nervous system involvement is a peculiar feature in our family since no other MERRF families without any central nervous system involvement have been reported to date. A possible explanation is that the mutational load remains low, especially in CNS, and prevents tissues/organs from reaching the pathologic threshold. The fact that this occurs throughout generations and that the tissue with the highest energetic demand is clinically spared, suggests that regulatory genes and/or pathways affect mitochondrial segregation and replication, and protect organs from progressive dysfunction. As suggested by Letrit et al., a lack of correlation between the degree of mtDNA heteroplasmy and clinical symptoms related to a particular organ can indicate the presence of tissue-specific nuclear factors that modify the phenotypic expression of the A8344G mutation, or, perhaps rather than a specific nuclear factor, there are merely tissue differences in the requirements for the particular subunit of the respiratory chain involved [
21].
6. Conclusions
In conclusion, our report highlights the broad clinical spectrum of MERRF syndrome, which can also occur as a pure myopathy. Given the large number of atypical cases, we would like to emphasize that it is easy to underestimate progressive and even potentially invalidating diseases.
The combination of a high clinical suspect, histological, molecular genetics, and biochemical investigation remains essential for the diagnosis of MERRF.
Author Contributions
M.S. and M.M. conducted clinical analysis; M.S., M.M. and G.P.C. collected clinical data; M.R., S.Z., L.N., D.R. and P.C. analyzed the data; M.R., S.Z. and M.M. wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was partially supported by the Italian Ministry of Health (Ministero della Salute Ricerca Corrente n° 245). This work was promoted within the European Reference Network (ERN) for Neuromuscular Diseases (M.S. as HCP Representative for the Italian ERN—NMD).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Ethics Committee of the IRCCS Ca’ Granda Foundation, Ospedale Maggiore Policlinico, Milano, Italy.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data for this article are not publicly available to ensure patient anonymity. Requests to access the data should be directed to the corresponding author.
Acknowledgments
We thank the Associazione Centro Dino Ferrari for its support. Muscle biopsy and DNA samples were provided by the Bank of muscle tissue, peripheral nerve, DNA, and Cell Culture, member of Telethon Network of Genetic biobanks, at IRCCS Ca’ Granda Foundation, Ospedale Maggiore Policlinico, Milano, Italy. This work was promoted within the European Reference Network (ERN) for Neuromuscular Diseases (M.S. as HCP Representative for the Italian ERN—NMD).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Zeviani, M.; Viscomi, C. Mitochondrial Neurodegeneration. Cells 2022, 11, 637. [Google Scholar] [CrossRef]
- Shoffner, J.M.; Lott, M.T.; Lezza, A.M.S.; Seibel, P.; Ballinger, S.W.; Wallace, D.C. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA-lys mutation. Cell 1990, 61, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, N.; Tokiguchi, S.; Shirakawa, K.; Tsubaki, T. Myoclonus epilepsy associated with ragged-red fibers (mitochondrial abnormalities): Disease entity or a syndrome? Light- and electron-microscopic studies of two cases and a review of the literature. J. Neurol. Sci. 1980, 47, 117–133. [Google Scholar] [CrossRef] [PubMed]
- Lorenzoni, P.J.; Scola, R.H.; Kay, C.S.; Arndt, R.C.; Silvado, C.E.; Werneck, L.C. MERRF: Clinical features, muscle biopsy and molecular genetics in Brazilian patients. Mitochondrion 2011, 11, 528–532. [Google Scholar] [CrossRef]
- Lombes, A.; Mendell, J.R.; Nakase, H.; Barohn, R.J.; Bonilla, E.; Zeviani, M.; Yates, A.J.; Omerza, J.; Gales, T.L.; Nakahara, K.; et al. Myoclonic epilepsy and ragged-red fibers with cytochrome oxidase deficiency: Neuropathology, biochemistry, and molecular genetics. Ann. Neurol. 1989, 26, 20–33. [Google Scholar] [CrossRef]
- Mancuso, M.; Petrozzi, L.; Filosto, M.; Nesti, C.; Rocchi, A.; Choub, A.; Pistolesi, S.; Massetani, R.; Fontanini, G.; Siciliano, G. MERRF syndrome without ragged-red fibers: The need for molecular diagnosis. Biochem. Biophys. Res. Commun. 2007, 354, 1058–1060. [Google Scholar] [CrossRef]
- Larsson, N.G.; Tulinius, M.H.; Holme, E.; Oldfors, A.; Andersen, O.; Wahlström, J.; Aasly, J. Segregation and manifestations of the mtDNA tRNA(Lys) A-->G(8344) mutation of myoclonus epilepsy and ragged-red fibers (MERRF) syndrome. Am. J. Hum. Genet. 1992, 51, 1201. [Google Scholar]
- Holme, E.; Larsson, N.G.; Oldfors, A.; Tulinius, M.; Sahlin, P.; Stenman, G. Multiple symmetric lipomas with high levels of mtDNA with the tRNA(Lys) A-->G(8344) mutation as the only manifestation of disease in a carrier of myoclonus epilepsy and ragged-red fibers (MERRF) syndrome. Am. J. Hum. Genet. 1993, 52, 551–556. [Google Scholar]
- Larsson, N.G.; Tulinius, M.H.; Holme, E.; Oldfors, A. Pathogenetic aspects of the A8344G mutation of mitochondrial DNA associated with MERRF syndrome and multiple symmetric lipomas. Muscle Nerve Suppl. 1995, 3, 102–106. [Google Scholar] [CrossRef]
- Muñoz-Málaga, A.; Bautista, J.; Salazar, J.A.; Aguilera, I.; Garcia, R.; Chinchon, I.; Segura, M.D.; Campos, Y.; Arenas, J. Lipomatosis, proximal myopathy, and the mitochondrial 8344 mutation. A lipid storage myopathy? Muscle Nerve 2000, 23, 538–542. [Google Scholar] [CrossRef]
- Silvestri, G.; Ciafaloni, E.; Santorelli, F.M.; Shanske, S.; Servidei, S.; Graf, W.D.; Sumi, M.; Di Mauro, S. Clinical features associated with the A-to-G transition at nucleotide 8344 of mtDNA (‘MERRF mutation’). Neurology 1993, 43, 1200–1206. [Google Scholar] [CrossRef]
- Hammans, S.R.; Sweeney, M.G.; Brockington, M.; Lennox, G.G.; Lawton, N.F.; Kennedy, C.R.; Morgan-Hughes, J.A.; Harding, A.E. The mitochondrial DNA transfer RNA-lys A-to-G (8344) mutation and the syndrome of myoclonic epilepsy with ragged red fibres (MERRF): Relationship of clinical phenotype to proportion of mutant mitochondrial DNA. Brain 1993, 116, 617–632. [Google Scholar] [CrossRef]
- Vallance, H.D.; Jeven, G.; Wallace, D.C.; Brown, M.D. A case of sporadic infantile histiocytoid cardiomyopathy caused by the A8344G (MERRF) mitochondrial DNA mutation. Pediatr. Cardiol. 2004, 25, 538–540. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Crumley, R.; Ringman, J.M. Spasmodic dysphonia in a patient with the A to G transition at nucleotide 8344 in mitochondrial DNA. Mov. Disord. 2003, 18, 716–718. [Google Scholar] [CrossRef]
- Horvath, R.; Kley, R.A.; Lochmuller, H.; Vorgerd, M. Parkinson syndrome, neuropathy, and myopathy caused by the mutation A8344G (MERRF) in tRNALys. Neurology 2007, 68, 56–58. [Google Scholar] [CrossRef] [PubMed]
- Orcesi, S.; Gorni, K.; Termine, C.; Uggetti, C.; Veggiotti, P.; Carrara, F.; Zeviani, M.; Berardinelli, A.; Lanzi, G. Bilateral putaminal necrosis associated with the mitochondrial DNA A8344G myoclonus epilepsy with ragged red fibers (MERRF) mutation: An infantile case. J. Child Neurol. 2006, 21, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, F.R.; Bartels, C.; Kirches, E.; Mawrin, C.; Wallesch, C.W. Unusual presentations of patients with the mitochondrial MERRF mutation A8344G. Clin. Neurol. Neurosurg. 2008, 110, 859–863. [Google Scholar] [CrossRef]
- Erol, I.; Alehan, F.; Horvath, R.; Schneiderat, P.; Talim, B. Demyelinating disease of central and peripheral nervous systems associated with a A8344G mutation in tRNALys. Neuromuscul. Disord. 2009, 19, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Vastagh, I.; Gál, A.; Reményi, V.; Semjén, J.; Lukács, T.; Valikovics, A.; Molnár, M.J. A8344G mutation of the mitochondrial DNA with typical mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes syndrome. Ideggyogy Sz. 2011, 64, 399–403. [Google Scholar]
- Lertrit, P.; Noer, A.S.; Byrne, E.; Marzuki, S. Tissue segregation of a heteroplasmic mtDNA mutation in MERRF (myoclonic epilepsy with ragged red fibers) encephalomyopathy. Hum. Genet. 1992, 90, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, G.; Focher, F.; Verri, A.; Spadari, S.; Banfi, P.; Gerosa, E.; Mazzarello, P. Myoclonus epilepsy and ragged-red fibers: Blood mitochondrial DNA heteroplasmy in affected and asymptomatic members of a family. Acta Neurol. Scand. 1993, 88, 406–409. [Google Scholar] [CrossRef]
- Ripolone, M.; Ronchi, D.; Violano, R.; Vallejo, D.; Fagiolari, G.; Barca, E.; Lucchini, V.; Colombo, I.; Villa, L.; Berardinelli, A.; et al. Impaired Muscle Mitochondrial Biogenesis and Myogenesis in Spinal Muscular Atrophy. JAMA Neurol. 2015, 72, 666–675. [Google Scholar] [CrossRef]
- Costanza, L.; Moggio, M. Muscular dystrophies: Histology, immunohistochemistry, molecular genetics and management. Curr. Pharm. Des. 2010, 16, 978–987. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, M.; Tanno, Y.; Horai, S.; Ozawa, T.; Miyatake, T.; Tsuji, S. A common mitochondrial DNA mutation in the tRNA-lys of patients with myoclonus epilepsy associated with ragged-red fibers. Biochem. Int. 1990, 21, 789–796. [Google Scholar] [PubMed]
- Silvestri, G.; Moraes, C.T.; Shanske, S.; Oh, S.J.; Di Mauro, S. A new mtDNA mutation in the tRNA-lys gene associated with myoclonic epilepsy and ragged-red fibers (MERRF). Am. J. Hum. Genet. 1992, 51, 1213–1217. [Google Scholar]
- Zeviani, M.; Muntoni, F.; Savarese, N.; Serra, G.; Tiranti, V.; Carrara, F.; Mariotti, C.; Di Donato, S. A MERRF/MELAS overlap syndrome associated with a new point mutation in the mitochondrial DNA tRNA-lys gene. Europ. J. Hum. Genet. 1993, 1, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Yabe, I.; Sudo, A.; Hosoki, K.; Yaguchi, H.; Saitoh, S.; Sasaki, H. MERRF/MELAS overlap syndrome: A double pathogenic mutation in mitochondrial tRNA genes. J. Med. Genet. 2010, 47, 659–664. [Google Scholar] [CrossRef] [Green Version]
- Shtilbans, A.; Shanske, S.; Goodman, S.; Sue, C.M.; Bruno, C.; Johnson, T.L.; Lava, N.S.; Waheed, N.; Di Mauro, S. G8363A mutation in the mitochondrial DNA transfer ribonucleic acid Lys gene: Another cause of Leigh syndrome. J. Child Neurol. 2000, 15, 759–761. [Google Scholar] [CrossRef]
- Virgilio, R.; Ronchi, D.; Bordoni, A.; Fassone, E.; Bonato, S.; Donadoni, C.; Torgano, G.; Moggio, M.; Corti, S.; Bresolin, N.; et al. Mitochondrial DNA G8363A mutation in the tRNA Lys gene: Clinical, biochemical and pathological study. J. Neurol. Sci. 2009, 281, 85–92. [Google Scholar] [CrossRef]
- Jeeva-Patel, T.; Freund, P.; Margolin, E.A. Lipomatosis and optic neuropathy clinches the diagnosis of myoclonic epilepsy with ragged red fibres (MERRF) syndrome. BMJ Case Rep. 2021, 14, e240463. [Google Scholar] [CrossRef]
- Tanji, K.; Gamez, G.; Cervera, C.; Mearin, F.; Ortega, A.; de la Torre, J.; Montoya, J.; Andreu, A.L.; Di Mauro, S.; Bonilla, E. The A8344G mutation in mitochondrial DNA associated with stroke-like episodes and gastrointestinal dysfunction. Acta Neuropathol 2003, 105, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Guallar, J.P.; Vilà, M.R.; Lopez-Gallardo, E.; Solano, A.; Domingo, J.C.; Gamez, J.; Pineda, M.; Capablo, J.L.; Domingo, P.; Andreu, A.L.; et al. Altered expression of master regulatory genes of adipogenesis in lipomas form patients bearing tRNA Lys point mutations in mitochondrial DNA. Mol. Genet. Metab. 2006, 89, 283–285. [Google Scholar] [CrossRef]
- Boulet, L.; Karpati, G.; Shoubridge, E.A. Distribution and threshold expression of the tRNA-lys mutation in skeletal muscle of patients with myoclonic epilepsy and ragged-red fibers (MERRF). Am. J. Hum. Genet. 1992, 51, 1187–1200. [Google Scholar] [PubMed]
- Di Mauro, S.; Hirano, M.; Kaufmann, P.; Tanji, K.; Sano, M.; Shungu, D.C.; Bonilla, E.; De Vivo, D.C. Clinical features and genetics of myoclonic epilepsy with ragged red fibers. Adv. Neurol. 2002, 89, 217–229. [Google Scholar] [PubMed]
- Mancuso, M.; Filosto, M.; Mootha, V.K.; Rocchi, A.; Pistolesi, S.; Murri, L.; DiMauro, S.; Siciliano, G. A novel mitochondrial tRNA-phe mutation causes MERRF syndrome. Neurology 2004, 62, 2119–2121. [Google Scholar] [CrossRef] [PubMed]
| Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).

 ,
, {kind=link}
