
The Genomic Profile Associated with Risk of Severe Forms of COVID-19 in Amazonian Native American Populations

 , , , ,
, , , ,  , , , , and
, , , , and
Abstract
:
1. Introduction
2. Materials and Methods
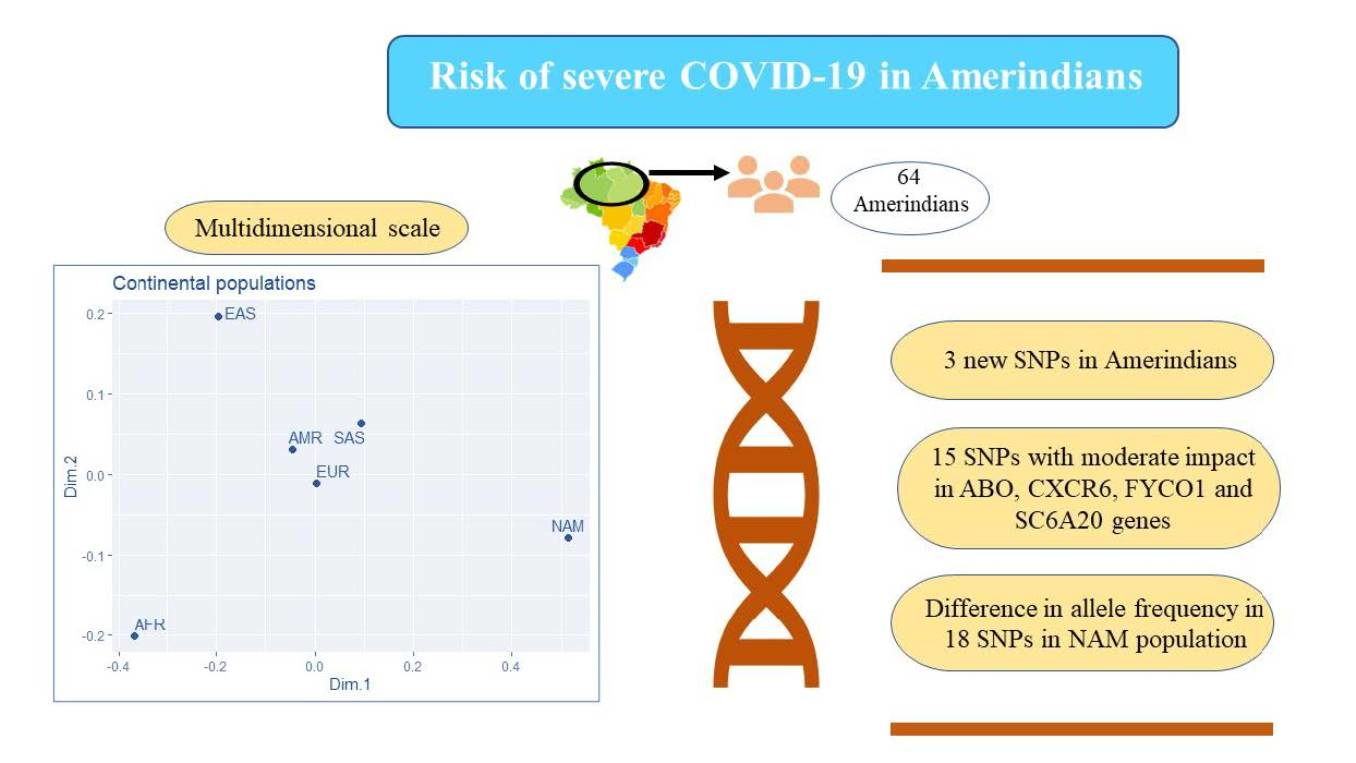
2.1. Study and Reference Populations
2.2. Extraction of DNA and Preparation of the Exome Library
2.3. Bioinformatic Analysis
2.4. Statistical Analyses
2.5. Selection of Variants
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, W.; Tang, J.; Wei, F. Updated understanding of the outbreak of 2019 novel coronavirus (2019-nCoV) in Wuhan, China. J. Med. Virol. 2020, 92, 441–447. [Google Scholar] [CrossRef] [Green Version]
- Cucinotta, D.; Vanelli, M. WHO Declares COVID-19 a Pandemic. Acta Bio-Med. Atenei Parm. 2020, 91, 157–160. [Google Scholar]
- World Health Organization. WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int (accessed on 6 February 2022).
- Samadizadeh, S.; Masoudi, M.; Rastegar, M.; Salimi, V.; Shahbaz, M.B.; Tahamtan, A. COVID-19: Why does disease severity vary among individuals? Respir. Med. 2021, 180, 106356. [Google Scholar] [CrossRef] [PubMed]
- Anastassopoulou, C.; Gkizarioti, Z.; Patrinos, G.P.; Tsakris, A. Human genetic factors associated with susceptibility to SARS-CoV-2 infection and COVID-19 disease severity. Hum. Genom. 2020, 14, 40. [Google Scholar] [CrossRef] [PubMed]
- Mario-Vásquez, J.E.; Naranjo-González, C.A.; Montiel, J.; Zuluaga, L.M.; Vásquez, A.M.; Tobón-Castaño, A.; Bedoya, G.; Segura, C. Association of variants in IL1B, TLR9, TREM1, IL10RA, and CD3G and Native American ancestry on malaria susceptibility in Colombian populations. Infect. Genet. Evol. 2021, 87, 104675. [Google Scholar] [CrossRef]
- Leal, D.F.D.V.B.; Da Silva, M.N.S.; Fernandes, D.C.R.D.O.; Rodrigues, J.C.G.; Barros, M.C.D.C.; Pinto, P.D.D.C.; Pastana, L.F.; Da Silva, C.A.; Fernandes, M.R.; De Assumpção, P.P.; et al. Amerindian genetic ancestry as a risk factor for tuberculosis in an amazonian population. PLoS ONE 2020, 15, e0236033. [Google Scholar] [CrossRef] [PubMed]
- Castro e Silva, M.A.; Ferraz, T.; Couto-Silva, C.M.; Lemes, R.B.; Nunes, K.; Comas, D.; Hünemeier, T. Population Histories and Genomic Diversity of South American Natives. Mol. Biol. Evol. 2021, 39, msab339. [Google Scholar] [CrossRef] [PubMed]
- Gilmour, B.; Alene, K.A.; Clements, A. The prevalence of tuberculosis and malaria in minority indigenous populations of South-East Asia and the Western Pacific Region: A systematic review and meta-analysis. Pathog. Glob. Health 2021, 1–19. [Google Scholar] [CrossRef]
- Pinto, P.; Salgado, C.; Santos, N.P.C.; Santos, S.; Ribeiro-dos-Santos, Â. Influence of Genetic Ancestry on INDEL Markers of NFKβ1, CASP8, PAR1, IL4 and CYP19A1 Genes in Leprosy Patients. PLoS Negl. Trop. Dis. 2015, 9, e0004050. [Google Scholar] [CrossRef] [Green Version]
- Tai, D.B.G.; Shah, A.; Doubeni, C.A.; Sia, I.G.; Wieland, M.L. The Disproportionate Impact of COVID-19 on Racial and Ethnic Minorities in the United States. Clin. Infect. Dis. 2021, 72, 703–706. [Google Scholar] [CrossRef]
- Da Cunha, A.A.; Corona, R.A.; Castilho-Martins, E.A. COVID-19 and race/color disparity: A brief analysis of the indigenous population in a state in the Brazilian Amazon. Einstein 2021, 19, eCE6734. [Google Scholar] [CrossRef]
- Stone, M.J.; Close, R.M.; Jentoft, C.K.; Pocock, K.; Lee-Gatewood, G.; Grow, B.I.; Parker, K.H.; Twarkins, A.; Nashio, J.T.; McAuley, J.B. High-Risk Outreach for COVID-19 Mortality Reduction in an Indigenous Community. Am. J. Public Health 2021, 111, 1939–1941. [Google Scholar] [CrossRef]
- Serrano-Coll, H.; Miller, H.; Rodríguez-Van, D.H.C.; Gastelbondo, B.; Novoa, W.; Oviedo, M.; Rivero, R.; Garay, E.; Mattar, S. High Prevalence of SARS-CoV-2 in an Indigenous Community of the Colombian Amazon Region. Trop. Med. Infect. Dis. 2021, 6, 191. [Google Scholar] [CrossRef]
- Cupertino, G.A.; Cupertino, M.D.C.; Gomes, A.P.; Braga, L.M.; Siqueira-Batista, R. COVID-19 and Brazilian Indigenous Populations. Am. J. Trop. Med. Hyg. 2020, 103, 609–612. [Google Scholar] [CrossRef]
- Secretaria de Saúde Indígena (SESAI). Boletim Epidemiológico 492, 2022, 1. Available online: https://www.gov.br/saude/pt-br/composicao/sesai (accessed on 30 November 2020).
- Lindenau, J.D.; Guimarães, L.S.P.; Friedrich, D.C.; Hurtado, A.M.; Hill, K.R.; Salzano, F.M.; Hutz, M.H. Cytokine gene polymorphisms are associated with susceptibility to tuberculosis in an Amerindian population. Int. J. Tuberc. Lung Dis. 2014, 18, 952–957. [Google Scholar] [CrossRef]
- Wang, S.; Lewis, C.M.; Jakobsson, M.; Ramachandran, S.; Ray, N.; Bedoya, G.; Rojas, W.; Parra-Marín, M.V.; A Molina, J.; Gallo, C.; et al. Genetic Variation and Population Structure in Native Americans. PLoS Genet. 2007, 3, e185. [Google Scholar] [CrossRef]
- Ellinghaus, D. Genomewide Association Study of Severe Covid-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534. [Google Scholar]
- Shelton, J.F.; Shastri, A.J.; Ye, C.; Weldon, C.H.; Filshtein-Sonmez, T.; Coker, D.; Symons, A.; Esparza-Gordillo, J.; Aslibekyan, S.; Auton, A.; et al. Trans-ancestry analysis reveals genetic and nongenetic associations with COVID-19 susceptibility and severity. Nat. Genet. 2021, 53, 801–808. [Google Scholar] [CrossRef]
- De Ramos, B.R.A.; D’Elia, M.P.B.; Amador, M.A.T.; Santos, N.P.C.; Santos, S.E.B.; da Castelli, E.C.; Witkin, S.S.; Miot, H.A.; Miot, L.D.B.; da Silva, M.G. Neither self-reported ethnicity nor declared family origin are reliable indicators of genomic ancestry. Genetica 2016, 144, 259–265. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, J.C.G.; de Souza, T.P.; Pastana, L.F.; Ribeiro dos Santos, A.M.; Fernandes, M.R.; Pinto, P.; Wanderley, A.V.; De Souza, S.J.; Kroll, J.E.; Pereira, A.L.; et al. Identification of NUDT15 gene variants in Amazonian Amerindians and admixed individuals from northern Brazil. PLoS ONE 2020, 15, e0231651. [Google Scholar] [CrossRef] [Green Version]
- Green, M.R.; Sambrook, J. Isolation of High-Molecular-Weight DNA Using Organic Solvents. Cold Spring Harb. Protoc. 2017, pdb.prot093450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro-dos-Santos, A.M.; Vidal, A.F.; Vinasco-Sandoval, T.; Guerreiro, J.; Santos, S.; Ribeiro-dos-Santos, Â. Exome Sequencing of Native Populations from the Amazon Reveals Patterns on the Peopling of South America. Front. Genet. 2020, 11, 548507. [Google Scholar] [CrossRef] [PubMed]
- Elhabyan, A.; Elyaacoub, S.; Sanad, E.; Abukhadra, A.; Elhabyan, A.; Dinu, V. The role of host genetics in susceptibility to severe viral infections in humans and insights into host genetics of severe COVID-19: A systematic review. Virus Res. 2020, 289, 198163. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camargo, S.M.R.; Vuille-dit-Bille, R.N.; Meier, C.F.; Verrey, F. ACE2 and gut amino acid transport. Clin. Sci. 2020, 134, 2823–2833. [Google Scholar] [CrossRef]
- Wei, Q.; Chen, Y.; Gu, Y.F.; Zhao, W. Molecular Characterization and Functional Analysis of Leucine Zipper Transcription Factor like 1 in Zebrafish (Danio rerio). Front. Physiol. 2019, 10, 801. [Google Scholar] [CrossRef]
- Downes, D.J.; Cross, A.R.; Hua, P.; Roberts, N.; Schwessinger, R.; Cutler, A.J.; Munis, A.M.; Brown, J.; Mielczarek, O.; de Andrea, C.E.; et al. Identification of LZTFL1 as a candidate effector gene at a COVID-19 risk locus. Nat. Genet. 2021, 53, 1606–1615. [Google Scholar] [CrossRef]
- Wu, X.; Sun, M.; Yang, Z.; Lu, C.; Wang, Q.; Wang, H.; Deng, C.; Liu, Y.; Yang, Y. The Roles of CCR9/CCL25 in Inflammation and Inflammation-Associated Diseases. Front. Cell Dev. Biol. 2021, 9, 2244. [Google Scholar] [CrossRef]
- Pathak, M.; Lal, G. The Regulatory Function of CCR9+ Dendritic Cells in Inflammation and Autoimmunity. Front. Imunol. 2020, 2219. [Google Scholar]
- Liu, G.; Abas, O.; Strickland, A.B.; Chen, Y.; Shi, M. CXCR6+CD4+ T cells promote mortality during Trypanosoma brucei infection. PLoS Pathog. 2021, 17, e1009968. [Google Scholar] [CrossRef]
- Luoma, A.M.; Suo, S.; Williams, H.L.; Sharova, T.; Sullivan, K.; Manos, M.; Bowling, P.; Hodi, F.S.; Rahma, O.; Sullivan, R.J.; et al. Molecular Pathways of Colon Inflammation Induced by Cancer Immunotherapy. Cell 2020, 182, 655–671.e22. [Google Scholar] [CrossRef]
- Balan, S.; Saxena, M.; Bhardwaj, N. Chapter One—Dendritic cell subsets and locations. In International Review of Cell and Molecular Biology; Lhuillier, C., Galluzzi, L., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 1–68. Available online: https://www.sciencedirect.com/science/article/pii/S193764481930067X (accessed on 30 November 2020).
- Lei, Y.; Takahama, Y. XCL1 and XCR1 in the immune system. Microbes Infect. 2012, 14, 262–267. [Google Scholar] [CrossRef]
- Pankiv, S.; Alemu, E.A.; Brech, A.; Bruun, J.-A.; Lamark, T.; Overvatn, A.; Bjørkøy, G.; Johansen, T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J. Cell Biol. 2010, 188, 253–269. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, C.; Menke, M.; Senger, F.; Mack, C.; Dierck, F.; Hille, S.; Schmidt, I.; Brunke, G.; Brünger, P.; Schmiedel, N.; et al. FYCO1 Regulates Cardiomyocyte Autophagy and Pre-vents Heart Failure Due to Pressure Overload In Vivo. JACC Basic Transl. Sci. 2021, 6, 365–380. [Google Scholar] [CrossRef]
- Iqbal, H.; Khan, S.Y.; Zhou, L.; Irum, B.; Ali, M.; Ahmed, M.R.; Shahzad, M.; Ali, M.H.; Naeem, M.A.; Riazuddin, S.; et al. Mutations in FYCO1 identified in families with congenital cataracts. Mol. Vis. 2020, 26, 334–344. [Google Scholar]
- Rothwell, S.; Lilleker, J.B.; Lamb, J.A. Genetics in inclusion body myositis. Curr. Opin. Rheumatol. 2017, 29, 639. [Google Scholar] [CrossRef]
- Franchini, M.; Crestani, S.; Frattini, F.; Sissa, C.; Bonfanti, C. ABO blood group and von Willebrand factor: Biological implications. Clin. Chem. Lab. Med. (CCLM) 2014, 52, 1273–1276. [Google Scholar] [CrossRef]
- Breiman, A.; Ruvën-Clouet, N.; Le Pendu, J. Harnessing the natural anti-glycan immune response to limit the transmission of enveloped viruses such as SARS-CoV-2. PLoS Pathog. 2020, 16, e1008556. [Google Scholar] [CrossRef]
- Goel, R.; Bloch, E.M.; Pirenne, F.; Al-Riyami, A.Z.; Crowe, E.; Dau, L.; Land, K.; Townsend, M.; Jecko, T.; Rahimi-Levene, N.; et al. ABO blood group and COVID-19: A review on behalf of the ISBT COVID-19 working group. Vox Sang. 2021, 116, 849–861. [Google Scholar] [CrossRef]
- Murray, G.P.; Post, S.R.; Post, G.R. ABO blood group is a determinant of von Willebrand factor protein levels in human pulmonary endothelial cells. J. Clin. Pathol. 2020, 73, 347–349. [Google Scholar] [CrossRef]
- IBGE. Censo Brasileiro de 2010. Rio de Janeiro-RJ. 2012. Available online: https://biblioteca.ibge.gov.br/visualizacao/periodicos/552/cd_2010_agsn_if.pdf (accessed on 30 November 2020).
- Salzano, F.M. Fatores dete rminísticos e estocásticos no processo microevolucionário humano. Actas V Congr. Latinoam. Genet. 1982, 1, 81–89. [Google Scholar]
- Amos, W.; Hoffman, J.I. Evidence that two main bottleneck events shaped modern human genetic diversity. Proc. Soc. Biol. Sci. 2010, 277, 131–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobbin, E.A.F.; Medeiros, J.A.G.; Costa, M.S.C.R.; Rodrigues, J.C.G.; Guerreiro, J.F.; Kroll, J.E.; Souza, S.; de Assumpção, P.; Ribeiro-Dos-Santos, Â.; Santos, S.; et al. Identification of Variants (rs11571707, rs144848, and rs11571769) in the BRCA2 Gene Associated with Hereditary Breast Cancer in Indigenous Populations of the Brazilian Amazon. Genes 2021, 12, 142. [Google Scholar] [CrossRef] [PubMed]
- O’Fallon, B.D.; Fehren-Schmitz, L. Native Americans experienced a strong population bottleneck coincident with European contact. Proc. Natl. Acad. Sci. USA 2011, 108, 20444–20448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Brito Vargas, L.; Beltrame, M.H.; Ho, B.; Marin, W.M.; Dandekar, R.; Montero-Martín, G.; Fernández-Viña, M.A.; Hurtado, A.M.; Hill, K.R.; Tsuneto, L.T.; et al. Remarkably Low KIR and HLA Diversity in Amerindians Reveals Signatures of Strong Purifying Selection Shaping the Centromeric KIR Region. Mol. Biol. Evol. 2022, 39, msab298. [Google Scholar] [CrossRef]
- Battilana, J.; Fagundes, N.J.R.; Heller, A.H.; Goldani, A.; Freitas, L.B.; Tarazona-Santos, E.; Munkhbat, B.; Munkhtuvshin, N.; Krylov, M.; Benevolenskaia, L.; et al. Alu insertion polymorphisms in Native Americans and related Asian populations. Ann. Hum. Biol. 2006, 33, 142–160. [Google Scholar] [CrossRef]
- Estrada-Mena, B.; Estrada, F.J.; Ulloa-Arvizu, R.; Guido, M.; Méndez, R.; Coral, R.; Canto, T.; Granados, J.; Rubí-Castellanos, R.; Rangel-Villalobos, H.; et al. Blood group O alleles in Native Americans: Implications in the peopling of the Americas. Am. J. Phys. Anthropol. 2010, 142, 85–94. [Google Scholar] [CrossRef]
- Salzano, F.M. The Amerindian Microcosm: Anthropology, Comparative History, Ecology, Genetics and Evolution; Hutz, M.H., Bortolini, M.C., Eds.; Cambridge Scholars Publishing: Newcastle upon Tyne, UK, 2019; p. 607. [Google Scholar]
- Hart, A.B.; Kranzler, H.R. Alcohol Dependence Genetics: Lessons Learned from Genome-Wide Association Studies (GWAS) and Post-GWAS Analyses. Alcohol. Clin. Exp. Res. 2015, 39, 1312–1327. [Google Scholar] [CrossRef] [Green Version]
- Nikpay, M.; Goel, A.; Won, H.-H.; Hall, L.M.; Willenborg, C.; Kanoni, S.; Saleheen, D.; Kyriakou, T.; Nelson, C.P.; Hopewell, J.C.; et al. A comprehensive 1000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 2015, 47, 1121–1130. [Google Scholar]
- Ko, D.C.; Urban, T.J. Understanding Human Variation in Infectious Disease Susceptibility through Clinical and Cellular GWAS. PLoS Pathog. 2013, 9, e1003424. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Wu, D.; Chen, H.; Yan, W.; Yang, D.; Chen, G.; Ma, K.; Xu, D.; Yu, H.; Wang, H.; et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: Retrospective study. BMJ 2020, 368, m1091. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Yang, Y.; Huang, H.; Li, D.; Gu, D.; Lu, X.; Zhang, Z.; Liu, L.; Liu, T.; Liu, Y.; et al. Relationship between the ABO Blood Group and the Coronavirus Disease 2019 (COVID-19) Susceptibility. Clin. Infect. Dis. 2021, 73, 328–331. [Google Scholar] [CrossRef]
- Halverson, M.S.; Bolnick, D.A. An ancient DNA test of a founder effect in native American ABO blood group frequencies. American J. Phys. Anthropol. 2008, 137, 342–347. [Google Scholar] [CrossRef]
- Fricke-Galindo, I.; Falfán-Valencia, R. Genetics Insight for COVID-19 Susceptibility and Severity: A Review. Front. Immunol. 2021, 12, 1057. [Google Scholar] [CrossRef]
- Cooling, L. Blood Groups in Infection and Host Susceptibility. Clin. Microbiol. Rev. 2015, 28, 801–870. [Google Scholar] [CrossRef] [Green Version]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Rigau, M.; Juan, D.; Valencia, A.; Rico, D. Intronic CNVs and gene expression variation in human populations. PLoS Genet. 2019, 15, e1007902. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Gene | Description * |
|---|---|
| SLC6A20 | This gene encodes the protein sodium–amino acid (proline) transporter 1 (SIT1), which interacts with the angiotensin-converting enzyme 2 (ACE2)—the SARS-CoV-2 cell-surface receptor—allowing its heterodimerization [19]. The heterodimerization of the ACE2 protein is necessary for the formation of a quaternary structure that functions as a binding site for the SARS-CoV-2 protein S [27]. |
| LZTFL1 | The LZTFL1 gene encodes the leucine zipper transcription factor-like 1, and its function is related to tumor-suppressor action and negative regulation of the hedgehog signaling pathways. This gene has high expression in lung tissues [25,28]; it is related to the functioning of the cilia of the pulmonary epithelium and to the signaling of important intracellular pathways, regulating the epithelial–mesenchymal transformation [29]. |
| CCR9 | CC chemokines are mainly responsible for the recruitment of lymphocytes. CCR9 is the receptor for the C-C chemokine ligand 25 (CCL25). The CCR9 receptor is mainly found on immature T lymphocytes and the surface of intestinal cells [30]. Animal studies have shown that the CCR9/CCL25 complex participates in the action of T helper 1 (Th1) cells. Another finding indicates that in knockout rats there was a reduction in the mRNA levels of pro-inflammatory cytokines (i.e., IL-6, IL-1β, and TNF-α) [30,31]. |
| CXCR6 | CXC chemokines have the highest ability to attract neutrophils and monocytes (30). CXCR6 is the receptor for CXCL16; in cellular studies and animal models, it has been shown to regulate inflammatory activity and influence the levels of INF-γ and TNF-α secreted by CD4+ T cells [32,33]. |
| XCR1 | XCR1 encodes the receptor of the XCL-1 ligand. The receptor triggers chemotactic signals in the presence of the ligand [34]. XCR1 is expressed in the lung tissue. Further reports suggest that XCL1 expression in NK cells and CD8+ T cells is constitutively detectable at a steady state, and is elevated during viral infection in mice and humans. The XCL1–XCR1 axis is important for efficient cytotoxic immune response mediated by CD8+ T cells [35]. |
| FYCO1 | This gene is responsible for the production of a Rab7 adapter protein, and has the function of assisting in the intracellular transport of autophagic vesicles via transport by microtubules. To carry out the transport, the encoded protein interacts with Rab7 GTPase, phosphatidylinositol-3-phosphate (PI3P), the autophagosome marker LC3, and the kinesin KIF5 [36,37]; it was previously found to be related to inclusion body myositis (IBM) and autosomal recessive congenital cataracts (CATC2) [38,39]. |
| ABO | The ABO gene encodes the enzyme alpha 1-3-galactosyltransferase, which transforms the H antigen expressed on the cell surface of several cell types into A and B antigens. Furthermore, the enzyme converts the H antigen into the von Willebrand factor [40]. Studies indicate that group A confers risk of developing severe forms of infection, while group O confers protection [1]. This effect is related to the expression of anti-A and anti-B antibodies that could neutralize the interaction of the virus protein S with ACE2, blocking its adsorption [41]. Another hypothesis would be its action in the formation of the von Willebrand factor and its relationship with its expression in the pulmonary endothelium, indirectly influencing pro-inflammatory regulation and cell adhesion [42,43]. |
| Gene | Position | SNP ID | Ref a | Var b | Impact Predicted by SnpEff | Variant Allele Frequency | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| NAM | AFR | AMR | EAS | EUR | SAS | ||||||
| ABO | 133256189 | rs55727303 | C | T | High | 0.281 | 0.001 | 0.108 | - | 0.012 | 0.026 |
| ABO | 133255902 | rs8176748 | C | T | Moderate | 0.814 | 0.243 | 0.431 | 0.290 | 0.221 | 0.220 |
| ABO | 133256085 | rs8176740 | A | T | Moderate | 0.814 | 0.242 | 0.431 | 0.289 | 0.221 | 0.220 |
| ABO | 133256264 | rs1053878 | G | A | Moderate | 0.016 | 0.256 | 0.068 | 0.151 | 0.099 | 0.029 |
| ABO | 133257465 | rs8176721 | G | A | Moderate | 0.000 | 0.155 | 0.017 | - | 0.005 | - |
| ABO | 133257486 | rs8176720 | T | C | Moderate | 0.814 | 0.489 | 0.494 | 0.483 | 0.337 | 0.464 |
| ABO | 133257246 | rs2073824 | A | G | Modifier | 0.728 | 0.469 | 0.496 | 0.549 | 0.336 | 0.465 |
| ABO | 133257320 | rs2073825 | A | T | Modifier | 0.235 | 0.241 | 0.431 | 0.289 | 0.222 | 0.225 |
| ABO | 133262062 | * | C | A | Modifier | 0.016 | - | - | - | - | - |
| ABO | 133275050 | rs616154 | C | T | Modifier | 0.031 | 0.531 | 0.408 | 0.408 | 0.535 | 0.631 |
| ABO | 133275068 | rs559723 | A | G | Modifier | 0.184 | 0.531 | 0.408 | 0.408 | 0.536 | 0.631 |
| CCR9 | 45894830 | rs7648467 | C | A | Modifier | 0.014 | 0.448 | 0.050 | - | 0.013 | 0.008 |
| CCR9 | 45897524 | rs17764980 | G | A | Modifier | 0.000 | 0.005 | 0.058 | 0.004 | 0.120 | 0.383 |
| CXCR6 | 45946488 | rs2234355 | G | A | Moderate | 0.033 | 0.491 | 0.068 | - | 0.005 | 0.001 |
| FYCO1 | 45959401 | * | G | A | Low | 0.019 | - | - | - | - | - |
| FYCO1 | 45923752 | rs35678722 | G | A | Moderate | 0.083 | 0.012 | 0.001 | - | - | - |
| FYCO1 | 45966331 | rs13079478 | G | T | Moderate | 0.070 | 0.005 | 0.059 | 0.004 | 0.122 | 0.360 |
| FYCO1 | 45966333 | rs13059238 | T | C | Moderate | 0.070 | 0.019 | 0.063 | 0.004 | 0.123 | 0.359 |
| FYCO1 | 45966722 | rs113517878 | C | T | Moderate | 0.083 | 0.003 | 0.003 | - | - | - |
| FYCO1 | 45967298 | rs3796375 | G | A | Moderate | 0.822 | 0.093 | 0.565 | 0.661 | 0.431 | 0.372 |
| FYCO1 | 45967995 | rs33910087 | G | A | Moderate | 0.070 | 0.017 | 0.059 | 0.005 | 0.122 | 0.359 |
| FYCO1 | 45968372 | rs3733100 | C | G | Moderate | 0.885 | 0.210 | 0.643 | 0.667 | 0.556 | 0.731 |
| FYCO1 | 45968585 | rs4683158 | C | T | Moderate | 0.994 | 0.982 | 0.914 | 1.000 | 0.801 | 0.922 |
| FYCO1 | 45979767 | rs1306733846 | C | T | Moderate | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| FYCO1 | 45923467 | rs6800954 | C | T | Modifier | 0.143 | 0.287 | 0.193 | 0.307 | 0.216 | 0.148 |
| FYCO1 | 45936407 | rs1873002 | T | C | Modifier | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 |
| FYCO1 | 45938385 | rs9875616 | G | A | Modifier | 0.859 | 0.914 | 0.850 | 0.954 | 0.746 | 0.879 |
| FYCO1 | 45959378 | rs13069079 | G | A | Modifier | 0.000 | 0.005 | 0.058 | 0.004 | 0.121 | 0.371 |
| FYCO1 | 45959571 | rs1532071 | G | A | Modifier | 0.908 | 0.260 | 0.614 | 0.652 | 0.529 | 0.732 |
| FYCO1 | 45959735 | rs76597151 | G | A | Modifier | 0.014 | 0.017 | 0.062 | 0.004 | 0.122 | 0.371 |
| FYCO1 | 45969944 | rs17214952 | A | G | Modifier | 0.014 | 0.019 | 0.063 | 0.004 | 0.123 | 0.360 |
| FYCO1 | 45973053 | rs41289622 | T | G | Modifier | 0.070 | 0.005 | 0.059 | 0.004 | 0.122 | 0.361 |
| FYCO1 | 45975359 | rs751552 | A | T | Modifier | 0.814 | 0.063 | 0.565 | 0.661 | 0.431 | 0.371 |
| FYCO1 | 45981341 | rs36122610 | G | A | Modifier | 0.054 | 0.005 | 0.059 | 0.004 | 0.122 | 0.358 |
| FYCO1 | 45984767 | rs3733097 | G | A | Modifier | 0.853 | 0.067 | 0.561 | 0.655 | 0.432 | 0.372 |
| LZTFL1 | 45828480 | rs1129183 | C | T | Moderate | 0.000 | 0.043 | 0.038 | 0.039 | 0.074 | 0.077 |
| LZTFL1 | 45827235 | * | TCTG | T | Modifier | 0.016 | - | - | - | - | - |
| LZTFL1 | 45842023 | rs138230559 | C | T | Modifier | 0.009 | 0.033 | - | - | - | - |
| LZTFL1 | 45842083 | rs141398338 | A | C | Modifier | 0.083 | 0.005 | - | - | 0.002 | - |
| SLC6A20 | 45759079 | rs140440513 | C | T | Moderate | 0.083 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| SLC6A20 | 45759901 | rs61731475 | T | C | Moderate | 0.000 | - | 0.006 | - | 0.014 | - |
| SLC6A20 | 45772602 | rs17279437 | G | A | Moderate | 0.017 | 0.005 | 0.043 | 0.006 | 0.092 | 0.031 |
| SLC6A20 | 45775922 | rs139429025 | T | C | Moderate | 0.000 | 0.012 | - | - | - | - |
| SLC6A20 | 45758379 | rs2251347 | C | T | Modifier | 0.994 | 0.990 | 0.976 | 0.921 | 0.954 | 0.972 |
| SLC6A20 | 45760066 | rs116638840 | C | T | Modifier | 0.027 | 0.076 | 0.010 | - | - | 0.003 |
| SLC6A20 | 45762899 | rs2191027 | C | T | Modifier | 0.014 | 0.020 | 0.193 | 0.017 | 0.299 | 0.149 |
| SLC6A20 | 45780132 | rs2252547 | T | C | Modifier | 0.155 | 0.465 | 0.614 | 0.450 | 0.591 | 0.516 |
| Gene | SNP ID | NAM vs. AFR * | NAM vs. AMR * | NAM vs. EAS * | NAM vs. EUR * | NAM vs. SAS * |
|---|---|---|---|---|---|---|
| ABO | rs55727303 | 1.66 × 10−19 | 1.08 × 10−3 | 1.40 × 10−17 | 6.75 × 10−14 | 8.82 × 10−11 |
| ABO | rs8176748 | 1.58 × 10−19 | 1.00 × 10−8 | 7.06 × 10−16 | 1.52 × 10−20 | 1.29 × 10−20 |
| ABO | rs8176740 | 1.58 × 10−19 | 1.00 × 10−8 | 7.06 × 10−16 | 1.52 × 10−20 | 1.29 × 10−20 |
| ABO | rs8176720 | 3.78 × 10−7 | 2.10 × 10−6 | 4.54 × 10−7 | 3.81 × 10−13 | 9.84 × 10−8 |
| ABO | rs1053878 | 6.04 × 10−7 | 0.149 | 0.001 | 0.020 | 1.000 |
| ABO | rs2073824 | 6.48 × 10−5 | 5.73 × 10−4 | 4.73 × 10−3 | 2.04 × 10−9 | 5.21 × 10−5 |
| ABO | rs559723 | 7.94 × 10−8 | 6.70 × 10−4 | 5.66 × 10−4 | 9.77 × 10−8 | 1.11 × 10−11 |
| ABO | rs616154 | 7.78 × 10−17 | 5.92 × 10−11 | 5.50 × 10−11 | 6.58 × 10−17 | 3.03 × 10−22 |
| ABO | rs2073825 | 1.000 | 3.0 × 10−3 | 0.382 | 0.874 | 0.874 |
| CCR9 | rs147314165 | 4.40 × 10−4 | 4.22 × 10−4 | 8.63 × 10−5 | 8.70 × 10−5 | 9.84 × 10−5 |
| CCR9 | rs7648467 | 2.45 × 10−14 | 0.330 | 0.113 | 0.570 | 0.461 |
| CXCR6 | rs2234355 | 5.37 × 10−15 | 0.398 | 0.035 | 0.100 | 0.037 |
| FYCO1 | rs3733100 | 4.13 × 10−28 | 3.67 × 10−5 | 1.45 × 10−4 | 5.81 × 10−8 | 5.27 × 10−3 |
| FYCO1 | rs3796375 | 2.35 × 10−37 | 4.62 × 10−5 | 6.62 × 10−3 | 1.20 × 10−9 | 3.18 × 10−12 |
| FYCO1 | rs35678722 | 3.39 × 10−3 | 4.22 × 10−4 | 8.63 × 10−5 | 8.70 × 10−5 | 9.84 × 10−5 |
| FYCO1 | rs113517878 | 8.44 × 10−5 | 4.22 × 10−4 | 8.63 × 10−5 | 8.70 × 10−5 | 9.84 × 10−5 |
| FYCO1 | rs4683158 | 0.614 | 0.008 | 1.000 | 2.49 × 10−6 | 0.015 |
| FYCO1 | rs13079478 | 4.40 × 10−4 | 0.578 | 2.76 × 10−4 | 0.410 | 1.51 × 10−6 |
| FYCO1 | rs13059238 | 0.012 | 0.591 | 2.76 × 10−4 | 0.410 | 1.51 × 10−6 |
| FYCO1 | rs33910087 | 0.009 | 0.578 | 0.001 | 0.410 | 1.51 × 10−6 |
| FYCO1 | rs1532071 | 6.14 × 10−25 | 1.36 × 10−6 | 1.32 × 10−5 | 8.95 × 10−10 | 1.82 × 10−3 |
| FYCO1 | rs3733097 | 2.08 × 10−45 | 2.84 × 10−6 | 6.14 × 10−4 | 2.84 × 10−11 | 7.34 × 10−14 |
| FYCO1 | rs751552 | 1.41 × 10−41 | 1.51 × 10−4 | 1.54 × 10−2 | 7.09 × 10−9 | 2.68 x10−11 |
| FYCO1 | rs1873002 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 |
| FYCO1 | rs9875616 | 0.168 | 1.000 | 6.0 x10−3 | 0.045 | 0.685 |
| FYCO1 | rs6800954 | 0.012 | 0.383 | 5.0 × 10−3 | 0.192 | 1.000 |
| FYCO1 | rs41289622 | 4.40 × 10−4 | 0.578 | 2.76 × 10−4 | 0.410 | 1.51 × 10−6 |
| FYCO1 | rs36122610 | 0.011 | 1.000 | 0.012 | 0.093 | 2.44 × 10−8 |
| FYCO1 | rs76597151 | 1.000 | 0.230 | 0.302 | 5.0 × 10−3 | 5.07 × 10−11 |
| FYCO1 | rs17214952 | 1.000 | 0.230 | 0.302 | 5.0 × 10−3 | 1.28 × 10−10 |
| LZTFL1 | rs141398338 | 4.40 × 10−4 | 4.22 × 10−4 | 8.63 × 10−5 | 8.70 × 10−5 | 9.84 × 10−5 |
| LZTFL1 | rs138230559 | 0.712 | 0.288 | 0.213 | 0.213 | 0.218 |
| SLC6A20 | rs140440513 | 2.59 × 10−5 | 4.22 × 10−4 | 8.63 × 10−5 | 8.70 × 10−5 | 9.84 × 10−5 |
| SLC6A20 | rs17279437 | 0.371 | 0.485 | 0.381 | 0.05 | 1.000 |
| SLC6A20 | rs2252547 | 1.08 × 10−14 | 6.54 × 10−12 | 3.18 × 10−6 | 2.12 × 10−11 | 1.95 × 10−8 |
| SLC6A20 | rs2251347 | 1.000 | 0.366 | 0.016 | 0.095 | 0.380 |
| SLC6A20 | rs116638840 | 0.306 | 0.236 | 0.035 | 0.035 | 0.068 |
| SLC6A20 | rs2191027 | 1.000 | 7.90 × 10−5 | 1.000 | 3.58 × 10−8 | 1.0 × 10−3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pastana, L.F.; Silva, T.A.; Gellen, L.P.A.; Vieira, G.M.; de Assunção, L.A.; Leitão, L.P.C.; da Silva, N.M.; Coelho, R.d.C.C.; de Alcântara, A.L.; Vinagre, L.W.M.S.; et al. The Genomic Profile Associated with Risk of Severe Forms of COVID-19 in Amazonian Native American Populations. J. Pers. Med. 2022, 12, 554. https://doi.org/10.3390/jpm12040554
Pastana LF, Silva TA, Gellen LPA, Vieira GM, de Assunção LA, Leitão LPC, da Silva NM, Coelho RdCC, de Alcântara AL, Vinagre LWMS, et al. The Genomic Profile Associated with Risk of Severe Forms of COVID-19 in Amazonian Native American Populations. Journal of Personalized Medicine. 2022; 12(4):554. https://doi.org/10.3390/jpm12040554
Chicago/Turabian StylePastana, Lucas Favacho, Thays Amâncio Silva, Laura Patrícia Albarello Gellen, Giovana Miranda Vieira, Letícia Almeida de Assunção, Luciana Pereira Colares Leitão, Natasha Monte da Silva, Rita de Cássia Calderaro Coelho, Angélica Leite de Alcântara, Lui Wallacy Morikawa Souza Vinagre, and et al. 2022. "The Genomic Profile Associated with Risk of Severe Forms of COVID-19 in Amazonian Native American Populations" Journal of Personalized Medicine 12, no. 4: 554. https://doi.org/10.3390/jpm12040554