
Next-Generation Sequencing in Lung Cancer Patients: A Comparative Approach in NSCLC and SCLC Mutational Landscapes

 , ,
, ,  , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. TCGA Datasets and Statistical Analysis
2.2. Patient Cohorts
2.3. Sample Processing and DNA Extraction
2.4. Sequencing Protocol and Data Analysis
2.5. Mutation Validation—SNP Genotyping Assay
3. Results
3.1. Genetic Alterations in the TCGA Datasets
3.2. Clinicopathological Characteristics of the Cohort
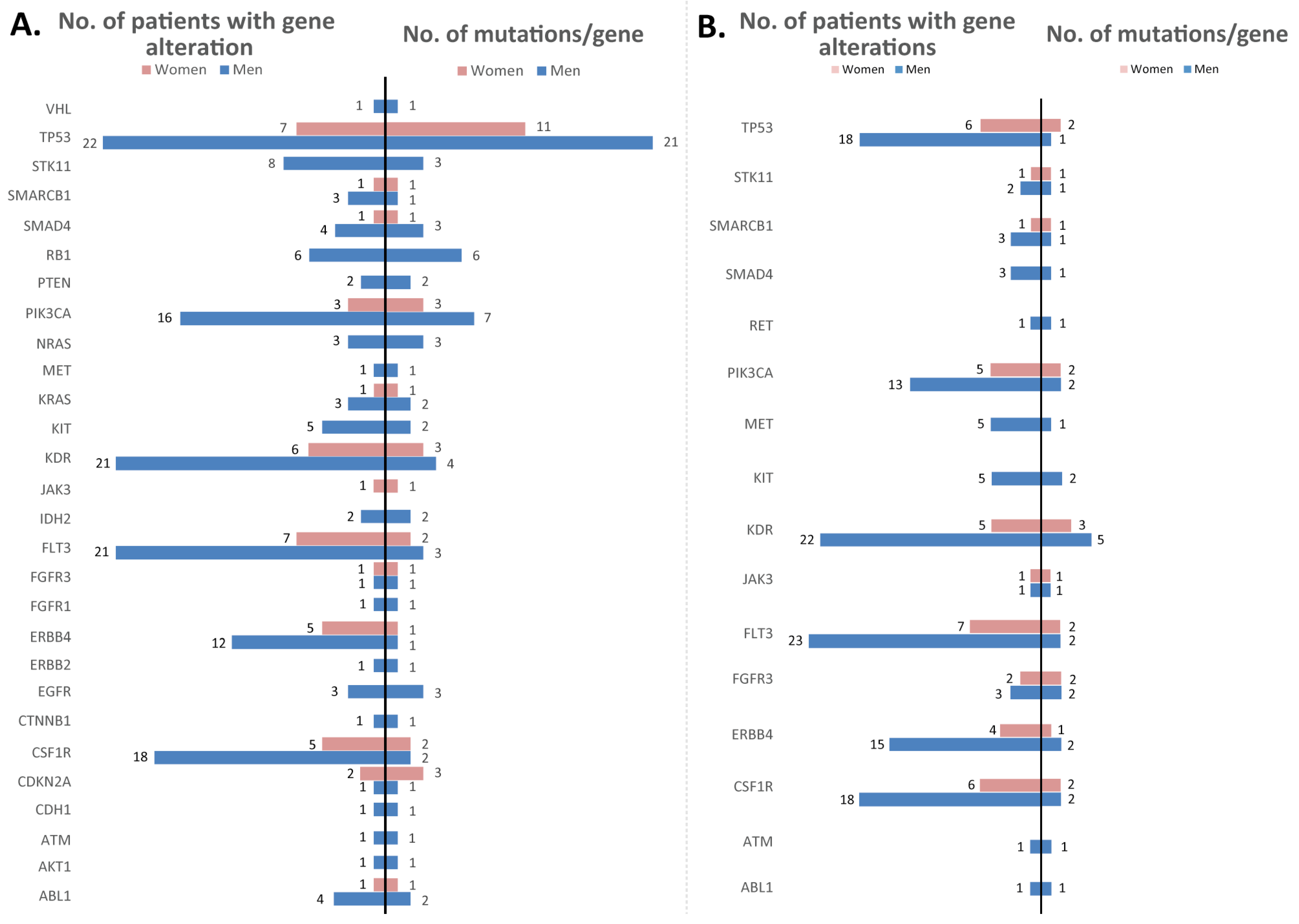
3.3. Mutations Identified in the Experimental Cohort
3.4. Mutation Validation
3.5. Statistical Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Kruglyak, K.M.; Lin, E.; Ong, F.S. Next-Generation Sequencing and Applications to the Diagnosis and Treatment of Lung Cancer. Adv. Exp. Med. Biol. 2016, 890, 123–136. [Google Scholar] [PubMed]
- National Lung Screening Trial Research Team; Aberle, D.R.; Adams, A.M.; Berg, C.D.; Black, W.C.; Clapp, J.D.; Fagerstrom, R.M.; Gareen, I.F.; Gatsonis, C.; Marcus, P.M.; et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N. Engl. J. Med. 2011, 365, 395–409. [Google Scholar]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karnes, H.E.; Duncavage, E.J.; Bernadt, C.T. Targeted next-generation sequencing using fine-needle aspirates from adenocarcinomas of the lung. Cancer Cytopathol. 2014, 122, 104–113. [Google Scholar] [CrossRef]
- Chen, Y.; Shi, J.-X.; Pan, X.-F.; Feng, J.; Zhao, H. Identification of candidate genes for lung cancer somatic mutation test kits. Genet. Mol. Biol. 2013, 36, 455–464. [Google Scholar] [CrossRef]
- Tuononen, K.; Maki-Nevala, S.; Sarhadi, V.K.; Wirtanen, A.; Ronty, M.; Salmenkivi, K.; Andrews, J.M.; Telaranta-Keerie, A.I.; Hannula, S.; Lagstrom, S.; et al. Comparison of targeted next-generation sequencing (NGS) and real-time PCR in the detection of EGFR, KRAS, and BRAF mutations on formalin-fixed, paraffin-embedded tumor material of non-small cell lung carcinoma-superiority of NGS. Genes Chromosomes Cancer 2013, 52, 503–511. [Google Scholar] [CrossRef]
- Buttitta, F.; Felicioni, L.; Del Grammastro, M.; Filice, G.; Di Lorito, A.; Malatesta, S.; Viola, P.; Centi, I.; D’Antuono, T.; Zappacosta, R.; et al. Effective Assessment of egfr Mutation Status in Bronchoalveolar Lavage and Pleural Fluids by Next-Generation Sequencing. Clin. Cancer Res. 2013, 19, 691–698. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.S.; Song, I.H.; Lee, A.; Kang, J.; Lee, Y.S.; Lee, I.K.; Song, Y.S.; Lee, S.H. Enhancing the landscape of colorectal cancer using targeted deep sequencing. Sci. Rep. 2021, 11, 8154. [Google Scholar] [CrossRef]
- Cai, H.; Jing, C.; Chang, X.; Ding, D.; Han, T.; Yang, J.; Lu, Z.; Hu, X.; Liu, Z.; Wang, J.; et al. Mutational landscape of gastric cancer and clinical application of genomic profiling based on target next-generation sequencing. J. Transl. Med. 2019, 17, 189. [Google Scholar] [CrossRef] [Green Version]
- Andrikopoulou, A.; Chatzinikolaou, S.; Kyriopoulos, I.; Bletsa, G.; Kaparelou, M.; Liontos, M.; Dimopoulos, M.-A.; Zagouri, F. The Mutational Landscape of Early-Onset Breast Cancer: A Next-Generation Sequencing Analysis. Front. Oncol. 2022, 11, 797505. [Google Scholar] [CrossRef] [PubMed]
- International HapMap Consortium. A haplotype map of the human genome. Nature 2005, 437, 1299–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kido, T.; Sikora-Wohlfeld, W.; Butte, A.J.; Kawashima, M.; Kikuchi, S.; Kamatani, N.; Patwardhan, A.; Chen, R.; Sirota, M.; Kodama, K.; et al. Are minor alleles more likely to be risk alleles? BMC Med. Genom. 2018, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Preusser, M.; Berghoff, A.S.; Koller, R.; Zielinski, C.C.; Hainfellner, J.A.; Liebmann-Reindl, S.; Popitsch, N.; Geier, C.B.; Streubel, B.; Birner, P. Spectrum of gene mutations detected by next generation exome sequencing in brain metastases of lung adenocarcinoma. Eur. J. Cancer 2015, 51, 1803–1811. [Google Scholar] [CrossRef]
- Takahashi, T.; Takahashi, T.; Suzuki, H.; Hida, T.; Sekido, Y.; Ariyoshi, Y.; Ueda, R. The p53 gene is very frequently mutated in small-cell lung cancer with a distinct nucleotide substitution pattern. Oncogene 1991, 6, 1775–1778. [Google Scholar]
- D’Amico, D.; Carbone, D.; Mitsudomi, T.; Nau, M.; Fedorko, J.; Russell, E.; Johnson, B.; Buchhagen, D.; Bodner, S.; Phelps, R. High frequency of somatically acquired p53 mutations in small-cell lung cancer cell lines and tumors. Oncogene 1992, 7, 339–346. [Google Scholar]
- Steels, E.; Paesmans, M.; Berghmans, T.; Branle, F.; Lemaitre, F.; Mascaux, C.; Meert, A.; Vallot, F.; Lafitte, J.; Sculier, J. Role of p53 as a prognostic factor for survival in lung cancer: A systematic review of the literature with a meta-analysis. Eur. Respir. J. 2001, 18, 705–719. [Google Scholar] [CrossRef] [Green Version]
- Baumann, M.; Zips, D.; Appold, S. Radiotherapy of lung cancer: Technology meets biology meets multidisciplinarity. Radiother. Oncol. 2009, 91, 279–281. [Google Scholar] [CrossRef]
- Viktorsson, K.; De Petris, L.; Lewensohn, R. The role of p53 in treatment responses of lung cancer. Biochem. Biophys. Res. Commun. 2005, 331, 868–880. [Google Scholar] [CrossRef]
- Grafone, T.; Palmisano, M.; Nicci, C.; Storti, S. An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: Biology and treatment. Oncol. Rev. 2012, 6, e8. [Google Scholar] [CrossRef] [Green Version]
- Stone, R.M.; DeAngelo, D.J.; Klimek, V.; Galinsky, I.; Estey, E.; Nimer, S.D.; Grandin, W.; Lebwohl, D.; Wang, Y.; Cohen, P.; et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood 2005, 105, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Borthakur, G.; Kantarjian, H.; Cortes, J.E.; Ravandi, F.; Zhang, W.; Konopleva, M.; Wright, J.J.; Faderl, S.; Verstovsek, S.; Mathews, S.; et al. Phase I study of sorafenib in patients with refractory or relapsed acute leukemias. Haematologica 2011, 96, 62–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, R.M.; Fischer, T.; Paquette, R.; Schiller, G.; Schiffer, C.A.; Ehninger, G.; Cortes, J.; Kantarjian, H.M.; DeAngelo, D.J.; Huntsman-Labed, A.; et al. Phase IB study of the FLT3 kinase inhibitor midostaurin with chemotherapy in younger newly diagnosed adult patients with acute myeloid leukemia. Leukemia 2012, 26, 2061–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jauhri, M.; Gupta, V.; Shokeen, Y.; Minhas, S.; Bhalla, S.; Aggarwal, S. KDR Mutation: A High-Frequency Rare Mutation and its Correlation with other Somatic Mutations in Indian Colorectal Cancer Patients. J. Next Gener. Seq. Appl. 2017, 4, 148. [Google Scholar] [CrossRef] [Green Version]
- Aredo, J.V.; Padda, S.K.; Kunder, C.A.; Han, S.S.; Neal, J.W.; Shrager, J.B.; Wakelee, H.A. Impact of KRAS mutation subtype and concurrent pathogenic mutations on non-small cell lung cancer outcomes. Lung Cancer 2019, 133, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Zito Marino, F.; Bianco, R.; Accardo, M.; Ronchi, A.; Cozzolino, I.; Morgillo, F.; Rossi, G.; Franco, R. Molecular heterogeneity in lung cancer: From mechanisms of origin to clinical implications. Int. J. Med. Sci. 2019, 16, 981–989. [Google Scholar] [CrossRef] [Green Version]
- Bonanno, S.; Zulato, E.; Pavan, A.; Attili, I.; Pasello, G.; Conte, P.; Indraccolo, S. LKB1, and Tumor Metabolism: The Interplay of Immune and Angiogenic Microenvironment in Lung Cancer. Int. J. Mol. Sci. 2019, 20, 1874. [Google Scholar] [CrossRef] [Green Version]
- Hua, X.; Zhao, W.; Song, L.; Sampson, J.; Wedge, D.C.; Shi, J.; Landi, M.T.; Pesatori, A.C.; Consonni, D.; Caporaso, N.E.; et al. Genetic and epigenetic intratumor heterogeneity impacts prognosis of lung adenocarcinoma. Nat. Commun. 2020, 11, 2459. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Training Cohort | Validation Cohort | |
|---|---|---|
| Characteristics | No. of Patients (%) | No. of Patients (%) |
| Total number | 32 | 32 |
| Age (years), median | 53–81 (62) | 42–81 (62) |
| Gender | ||
| Male | 25 (78.2) | 23 (71.9) |
| Female | 7 (21.8) | 9 (28.1) |
| pT stage | ||
| pT1 | 1 (3.1) | - |
| pT2 | 4 (12.5) | - |
| pT3 | 10 (31.2) | 7 (21.8) |
| pT4 | 17 (53.1) | 25 (78.2) |
| pN stage | ||
| pN0 | 2 (6.2) | 2 (6.2) |
| pN1 | - | 2 (6.2) |
| pN2 | 20 (62.5) | 22 (68.7) |
| pN3 | 10 (31.2) | 6 (18.7) |
| pM stage | ||
| pM0 | 14 (43.7) | 13 (40.6) |
| pM1 | 18 (56.3) | 19 (59.4) |
| Stage | ||
| IIIB | 14 (43.8) | 11 (34.4) |
| IIIC | 1 (3.1) | 2 (6.2) |
| IV | 17 (53.1) | 19 (59.4) |
| Histological type | ||
| NSCLC | 16 (50) | 16 (50) |
| SCLC | 16 (50) | 16 (50) |
| Smoking status | ||
| Active smoker | 16 (50) | 16 (50) |
| Former smoker | 13 (40.6) | 13 (40.6) |
| Never smoker | 3 (9.4) | 3 (9.4) |
| Training Cohort | ||||||||
|---|---|---|---|---|---|---|---|---|
| Patient | Sex | Age at Diagnosis | TNM | Stage | Histological Type | Active Smoker | Former Smoker | Days to Event |
| P8 | F | 58 | T3N2M0 | IIIB | adenocarcinoma | yes | yes | 116 |
| P12 | M | 67 | T3N2M0 | IIIB | SCLC | yes | yes | 41 |
| P13 | M | 66 | T3N3M0 | IIIB | adenocarcinoma | no | yes | 518 |
| P15 | M | 58 | T4N2M0 | IIIB | squamous cell carcinoma | no | yes | 311 |
| P16 | F | 76 | T4N3M0 | IIIC | squamous cell carcinoma | yes | yes | 584 |
| P19 | F | 67 | T3N3M1 | IV | adenocarcinoma | no | no | 871 |
| P20 | F | 81 | T4N2M0 | IIIB | adenocarcinoma | no | yes | 32 |
| P22 | M | 58 | T2N2M0 | IIIB | SCLC | yes | yes | 261 |
| P23 | M | 60 | T3N2M0 | IIIB | SCLC | no | yes | 336 |
| P24 | M | 60 | T4N2M1 | IV | SCLC | no | yes | 111 |
| P31 | M | 61 | T4N2M1 | IV | squamous cell carcinoma | no | yes | |
| P34 | M | 62 | T3N2M1 | IV | adenocarcinoma | yes | yes | 131 |
| P36 | M | 65 | T3N2M0 | IIIB | squamous cell carcinoma | no | yes | 934 |
| P37 | F | 63 | T3N3M1 | IV | SCLC | no | no | 408 |
| P38 | M | 77 | T4N3M1 | IV | SCLC | no | yes | 44 |
| P42 | M | 65 | T4N3M1 | IIIB | SCLC | yes | yes | |
| P50 | M | 62 | T4N3M1 | IV | squamous cell carcinoma | yes | yes | 162 |
| P63 | M | 66 | T4N2M1 | IV | adenocarcinoma | no | yes | |
| P66 | M | 62 | T4N2M1 | IV | SCLC | yes | yes | 62 |
| P68 | F | 58 | T3N2M1 | IV | adenocarcinoma | no | no | 582 |
| P73 | M | 65 | T4N3M0 | IIIB | SCLC | yes | yes | 116 |
| P75 | M | 61 | T4N2M0 | IIIB | SCLC | no | yes | 338 |
| P77 | M | 65 | T3N2M1 | IV | SCLC | no | yes | |
| P78 | M | 67 | T4N2M0 | IIIB | SCLC | no | yes | 250 |
| P80 | M | 67 | T2N2M1 | IV | squamous cell carcinoma | yes | yes | 33 |
| P82 | M | 58 | T4N0M0 | IIIB | squamous cell carcinoma | yes | yes | 32 |
| P84 | M | 60 | T2N2M1 | IV | SCLC | yes | yes | 227 |
| P85 | F | 53 | T4N2M0 | IIIB | squamous cell carcinoma | yes | yes | 70 |
| P86 | M | 71 | T2N0M1 | IV | adenocarcinoma | yes | yes | 163 |
| P88 | M | 59 | T4N3M1 | IV | SCLC | yes | yes | |
| P92 | M | 57 | T1N3M1 | IV | SCLC | yes | yes | |
| P107 | M | 56 | T4N2M1 | IV | SCLC | no | yes | 71 |
| Validation Cohort | ||||||||
| Patient | Sex | Age at Diagnosis | TNM | Stage | Histological Type | Active Smoker | Former Smoker | Days to Event |
| P110 | M | 55 | T3N2M1 | IV | squamous cell carcinoma | yes | yes | |
| P118 | M | 70 | T3N2M1 | IV | SCLC | no | yes | 46 |
| P120 | M | 71 | T4N2M0 | IIIB | SCLC | yes | yes | 185 |
| P122 | M | 58 | T4N0M1 | IV | adenocarcinoma | no | yes | 450 |
| P124 | M | 61 | T4N3M1 | IV | SCLC | yes | yes | 47 |
| P129 | M | 60 | T4N2M0 | IIIB | squamous cell carcinoma | yes | yes | 16 |
| P130 | F | 68 | T3N2M1 | IV | adenocarcinoma | no | yes | 143 |
| P133 | F | 56 | T4N2M1 | IV | SCLC | yes | yes | 213 |
| P136 | F | 67 | T3N2M0 | IIIB | squamous cell carcinoma | no | yes | 73 |
| P137 | F | 50 | T4N2M0 | IIIB | adenocarcinoma | no | yes | 286 |
| P138 | M | 62 | T4N1M1 | IV | adenocarcinoma | no | no | 231 |
| P139 | M | 57 | T4N2M0 | IIIB | SCLC | no | yes | 489 |
| P140 | M | 69 | T4N2M1 | IV | SCLC | no | yes | 3 |
| P144 | M | 75 | T4N3M1 | IV | SCLC | no | yes | 622 |
| P149 | M | 81 | T4N2M1 | IV | SCLC | yes | yes | 816 |
| P150 | M | 60 | T3N1M1 | IV | SCLC | no | yes | 304 |
| P153 | M | 61 | T4N3M0 | IIIC | adenocarcinoma | no | no | |
| P159 | M | 62 | T4N2M0 | IIIB | squamous cell carcinoma | yes | yes | 359 |
| P160 | M | 80 | T4N2M1 | IV | squamous cell carcinoma | yes | no | 888 |
| P161 | F | 58 | T4N3M1 | IV | SCLC | yes | yes | 302 |
| P162 | F | 53 | T4N2M1 | IV | SCLC | yes | yes | 887 |
| P163 | F | 68 | T4N2M0 | IIIB | adenocarcinoma | no | yes | 884 |
| P164 | M | 57 | T4N2M0 | IIIB | SCLC | no | yes | 883 |
| P166 | M | 50 | T4N2M1 | IV | adenocarcinoma | no | yes | 65 |
| P170 | M | 75 | T4N3M1 | IV | squamous cell carcinoma | no | no | 59 |
| P171 | M | 56 | T4N3M0 | IIIC | SCLC | yes | yes | 153 |
| P181 | M | 73 | T4N1M1 | IV | SCLC | no | yes | 278 |
| P184 | M | 42 | T4N2M0 | IIIB | SCLC | yes | yes | 23 |
| P186 | F | 78 | T4N2M1 | IV | squamous cell carcinoma | yes | yes | 8 |
| P191 | M | 75 | T3N2M0 | IIIB | adenocarcinoma | yes | yes | 812 |
| P193 | M | 68 | T3N2M0 | IIIB | squamous cell carcinoma | yes | yes | 319 |
| P194 | F | 64 | T4N2M1 | IV | SCLC | yes | yes | |
| Gene | No. Pathogenic Mutations/Gene | Mutation | Amino Acid Change | NSCLC | SCLC (n = 16) | Exon | Variance | Sample | |
|---|---|---|---|---|---|---|---|---|---|
| LUAD (n = 8) | LUSC (n = 8) | TT/Blood | |||||||
| ABL1 | 1 | c.992A>G | p.Asn331Ser | - | - | 1 | 6 | Missense | 1/1 |
| CDKN2A | 1 | c.220G>T | p.Asp74Tyr | - | 1 | - | 2 | Missense | 1/0 |
| CTNNB1 | 1 | c.136C>G | p.Leu46Val | 1 | - | - | 3 | Missense | 1/0 |
| ERBB2 | 1 | c.2329G>A | p.Val777Met | - | - | 1 | 20 | Missense | 1/0 |
| IDH2 | 1 | c.419G>A | p.Arg140Gln | 1 | - | - | 4 | Missense | 1/0 |
| KIT | 2 | c.1621A>C | p.Met541Leu | 1 | 1 | 2 | 10 | Missense | 1/1 |
| c.1588G>A | p.Val530Ile | 1 | - | - | 10 | Missense | 0/1 | ||
| NRAS | 2 | c.182A>G | p.Gln61Arg | - | 1 | - | 3 | Missense | 1/0 |
| c.182A>T | p.Arg140Gln | - | - | 1 | 3 | Missense | 1/0 | ||
| PIK3CA | 3 | c.1624G>A | p.Glu542Lys | - | - | 2 | 10 | Missense | 1/0 |
| c.3196G>A | p.Ala1066Thr | 1 | 1 | 4 | 21 | Missense | 1/0 | ||
| c.328G>A | p.Glu110Lys | - | - | 1 | 2 | Missense | 1/0 | ||
| PTEN | 1 | c.388C>T | p.Arg130Ter | - | - | 1 | 5 | Nonsense | 1/0 |
| RB1 | 1 | c.409G>T | p.Glu137Ter | - | - | 1 | 4 | Nonsense | 1/0 |
| RET | 1 | c.409G>T | p.Ser649Leu | - | - | 1 | 11 | Missense | 0/1 |
| SMAD4 | 2 | c.546C>G | p.Ile182Met | - | - | 1 | 5 | Missense | 1/0 |
| c.1081C>T | p.Arg361Cys | - | - | 1 | 9 | Missense | 1/0 | ||
| STK11 | 1 | c.465-1G>T | - | - | - | 1 | 4 | Unknown | 1/0 |
| TP53 | 23 | c.1000G>T | p.Gly334Trp | 1 | - | - | 10 | Missense | 1/0 |
| c.1001G>T | p.Gly334Val | 1 | - | - | 10 | Missense | 1/0 | ||
| c.1036G>T | p.Glu346Ter | - | - | 1 | 10 | Nonsense | 1/0 | ||
| c.215C>G | p.Pro72Arg | 5 | 4 | 12 | 4 | Missense | 1/1 | ||
| c.400T>A | p.Phe134Ile | - | - | 1 | 5 | Missense | 1/0 | ||
| c.461G>T | p.Gly154Val | - | - | 1 | 5 | Missense | 1/0 | ||
| c.472C>G | p.Arg158Gly | - | - | 1 | 5 | Missense | 1/0 | ||
| c.473G>T | p.Arg158Leu | - | 1 | - | 5 | Missense | 1/0 | ||
| c.500A>G | p.Gln167Arg | 1 | - | - | 5 | Missense | 0/1 | ||
| c.514G>T | p.Val172Phe | - | 1 | - | 5 | Missense | 1/0 | ||
| c.559+1G>T | - | 1 | - | - | 5 | unknown | 1/0 | ||
| c.575A>G | p.Gln192Arg | - | 1 | - | 6 | Missense | 1/0 | ||
| c.713G>T | p.Cys238Phe | - | - | 1 | 7 | Missense | 1/0 | ||
| c.722C>T | p.Ser241Phe | - | - | 1 | 7 | Missense | 1/0 | ||
| c.725G>T | p.Cys242Phe | - | 1 | - | 7 | Missense | 1/0 | ||
| c.742C>T | p.Arg248Trp | - | - | 1 | 7 | Missense | 1/0 | ||
| c.797G>T | p.Gly266Val | - | - | 1 | 8 | Missense | 1/0 | ||
| c.817C>A | p.Arg273Ser | 1 | - | - | 8 | Missense | 1/0 | ||
| c.832C>T | p.Pro278Ser | - | - | 1 | 8 | Missense | 1/0 | ||
| c.850A>G | p.Thr284Ala | 1 | - | - | 8 | Missense | 1/0 | ||
| c.856G>A | p.Glu286Lys | - | 1 | - | 8 | Missense | 1/0 | ||
| c.872A>G | p.Lys291Arg | - | - | 1 | 8 | Missense | 1/0 | ||
| c.880G>T | p.Glu294Ter | - | 1 | 1 | 8 | Nonsense | 1/0 | ||
| Datasets TCGA | LUAD | LUSC | SCLC | ||||
|---|---|---|---|---|---|---|---|
| Gene | Altered/Unaltered | Logrank Test p-Value | Altered/Unaltered | Logrank Test p-Value | Altered/Unaltered | Logrank Test p-Value | |
| ABL1 | 3/218 | 0.590 | 5/170 | 0.286 | 1/109 | 0.729 | |
| CDKN2A | 55/166 | 0.0576 | 79/96 | 0.0531 | 1/109 | 0.359 | |
| CTNNB1 | 9/212 | 0.791 | 5/170 | 0.140 | 0/110 | - | |
| ERBB2 | 12/209 | 0.835 | 9/166 | 0.977 | 1/109 | 0.240 | |
| IDH2 | 4/217 | 0.0243 | 5/170 | 0.266 | 0/110 | - | |
| KIT | 9/212 | 0.976 | 15/160 | 0.360 | 7/103 | 0.528 | |
| NRAS | 7/214 | 4.558 × 10−4 | 6/169 | 0.122 | 0/110 | - | |
| PIK3CA | 19/202 | 0.793 | 105/70 | 0.938 | 3/107 | 0.574 | |
| PTEN | 6/215 | 0.641 | 30/145 | 0.283 | 7/103 | 0.441 | |
| RB1 | 17/204 | 0.910 | 18/157 | 0.418 | 80/30 | 0.410 | |
| SMAD4 | 11/210 | 0.480 | 10/165 | 0.882 | 2/108 | 0.112 | |
| STK11 | 42/179 | 0.412 | 6/169 | 0.0403 | 1/109 | 0.0272 | |
| TP53 | 103/118 | 0.173 | 142/33 | 0.0419 | 94/16 | 0.705 | |
| Altered TP53 | Altered KIT | Altered PIK3CA | Altered STK11 | Stage | Tumor Size | Lymph Node Metastasis | Distant Metastatic Spread | Adenocarcinoma | Squamous Cell Carcinoma | Small-Cell Lung Cancer | Death (Days) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| altered TP53 | 0.138409 | 0.148581 | 0.185695 | 0.127563 | 0.267435 | −0.06459 | 0.148581 | −0.0619 | −0.0619 | 0.107211 | 0.093676 | |
| altered KIT | 0.1384091 | 0.03253 | −0.04969 | 0.074796 | −0.00678 | 0.046953 | 0.060351 | 0.019821 | 0.019821 | −0.03452 | 0.049947 | |
| altered PIK3CA | 0.1485808 | 0.03253 | 0.218218 | 0.055228 | 0.217574 | 0.22771 | −0.01587 | −0.50918 | 0.218218 | 0.251976 | −0.03286 | |
| altered STK11 | 0.1856953 | −0.04969 | 0.218218 | 0.108465 | 0.110783 | 0.248452 | 0.218218 | 0 | −0.16667 | 0.144338 | 0.056454 | |
| Stage | 0.1275627 | 0.074796 | 0.055228 | 0.108465 | −0.10394 | −0.04296 | 0.96862 | 0 | −0.14548 | 0.125988 | −0.00042 | |
| Tumor size | 0.2674354 | −0.00678 | 0.217574 | 0.110783 | −0.10394 | 0.06877 | −0.08411 | −0.10585 | 0.052926 | 0.045835 | −0.04025 | |
| Lymph node metastasis | −0.06459093 | 0.046953 | 0.22771 | 0.248452 | −0.04296 | 0.06877 | −0.00216 | −0.19687 | −0.03937 | 0.204598 | 0.113633 | |
| Distant metastatic spread | 0.1485808 | 0.060351 | −0.01587 | 0.218218 | 0.96862 | −0.08411 | −0.00216 | −0.01827 | −0.1644 | 0.158193 | −0.00042 | |
| Adenocarcinoma | −0.06189845 | 0.019821 | −0.50918 | 0 | 0 | −0.10585 | −0.19687 | −0.01827 | −0.33333 | −0.57735 | 0.081326 | |
| Squamous cell carcinoma | −0.06189845 | 0.019821 | 0.218218 | −0.16667 | −0.14548 | 0.052926 | −0.03937 | −0.1644 | −0.33333 | −0.57735 | −0.0643 | |
| Small-cell lung cancer | 0.1072113 | −0.03452 | 0.251976 | 0.144338 | 0.125988 | 0.045835 | 0.204598 | 0.158193 | −0.57735 | −0.57735 | −0.01636 | |
| Death (days) | 0.09367601 | 0.049947 | −0.03286 | 0.056454 | −0.00042 | −0.04025 | 0.113633 | −0.00042 | 0.081326 | −0.0643 | −0.01636 | |
| deceased | −0.01491 | 0.085205 | −0.10635 | −0.05723 | 0.085205 | −0.09343 | 0.036155 | 0.051473 | −0.63572 | |||
| Active smoker | −0.1072113 | −0.20179 | 0 | 0.144338 | −0.06299 | −0.1375 | 0.068199 | −0.03164 | −0.28868 | 0.216506 | 0.0625 | −0.24329 |
| Former smoker | −0.1034483 | −0.13566 | 0.364698 | 0.185695 | −0.1756 | 0.029484 | −0.18279 | −0.16621 | −0.30949 | 0.061898 | 0.214423 | −0.14787 |
| never smoker | 0.1034483 | 0.135656 | −0.3647 | −0.1857 | 0.175596 | −0.02948 | 0.182794 | 0.166209 | 0.309492 | −0.0619 | −0.21442 | 0.147874 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pop-Bica, C.; Ciocan, C.A.; Braicu, C.; Haranguș, A.; Simon, M.; Nutu, A.; Pop, L.A.; Slaby, O.; Atanasov, A.G.; Pirlog, R.; et al. Next-Generation Sequencing in Lung Cancer Patients: A Comparative Approach in NSCLC and SCLC Mutational Landscapes. J. Pers. Med. 2022, 12, 453. https://doi.org/10.3390/jpm12030453
Pop-Bica C, Ciocan CA, Braicu C, Haranguș A, Simon M, Nutu A, Pop LA, Slaby O, Atanasov AG, Pirlog R, et al. Next-Generation Sequencing in Lung Cancer Patients: A Comparative Approach in NSCLC and SCLC Mutational Landscapes. Journal of Personalized Medicine. 2022; 12(3):453. https://doi.org/10.3390/jpm12030453
Chicago/Turabian StylePop-Bica, Cecilia, Cristina Alexandra Ciocan, Cornelia Braicu, Antonia Haranguș, Marioara Simon, Andreea Nutu, Laura Ancuta Pop, Ondrej Slaby, Atanas G. Atanasov, Radu Pirlog, and et al. 2022. "Next-Generation Sequencing in Lung Cancer Patients: A Comparative Approach in NSCLC and SCLC Mutational Landscapes" Journal of Personalized Medicine 12, no. 3: 453. https://doi.org/10.3390/jpm12030453