
Fanconi Anemia Pathway in Colorectal Cancer: A Novel Opportunity for Diagnosis, Prognosis and Therapy

 ,
,  ,
,
Abstract
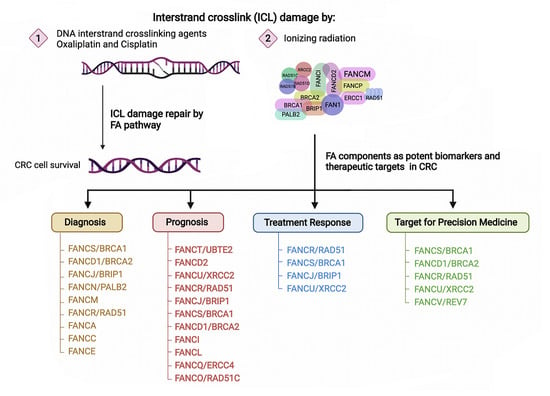
:
1. Introduction
2. The Fanconi Anemia Pathway in DNA Repair and Maintenance of Genome Integrity
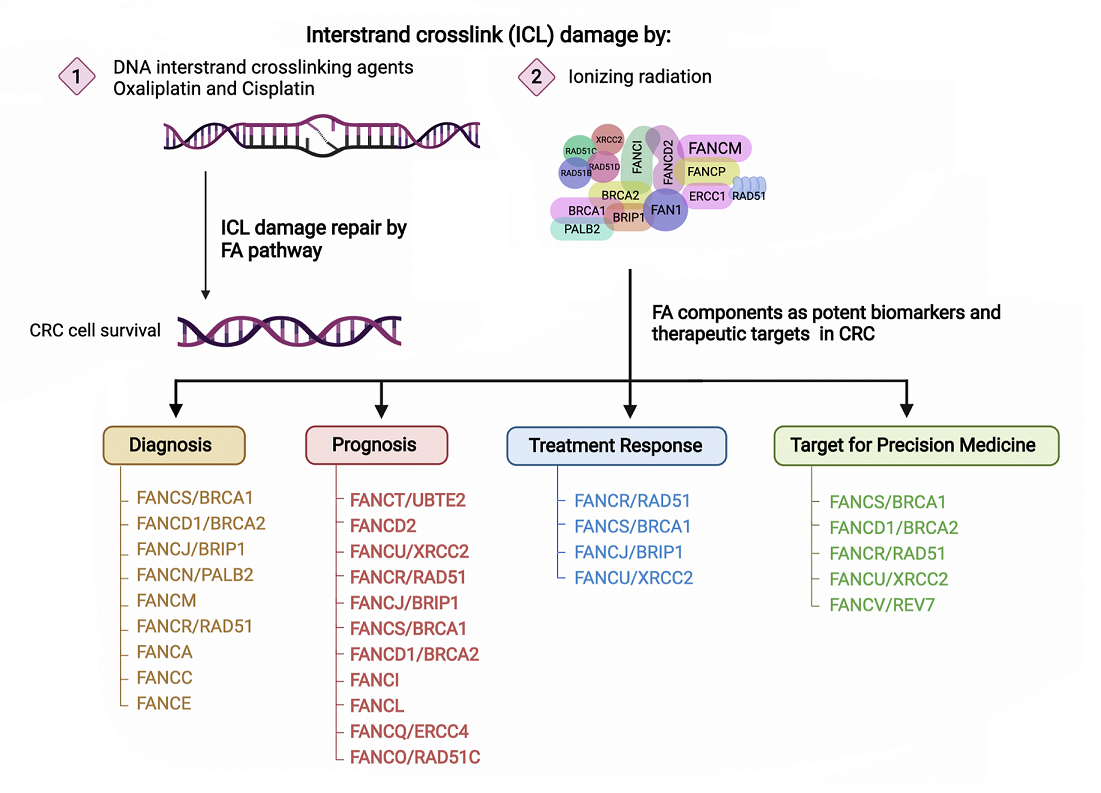
2.1. The FA Pathway and Inter-Strand Crosslink Repair
2.2. FA Proteins Stabilize Stalled Replication Forks
3. Potential Role of FANC Gene Mutations in Colorectal Cancer Susceptibility
3.1. Germline Monoallelic Mutations
3.2. Somatic Mutations
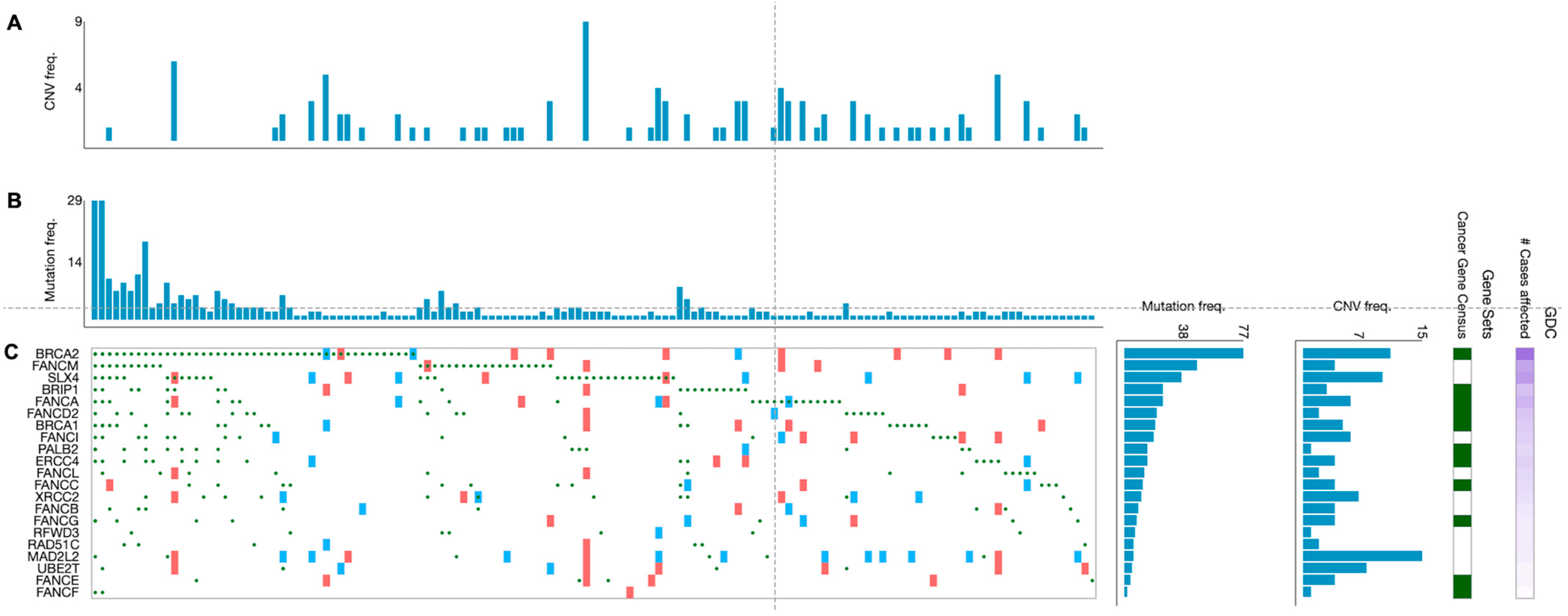
4. FA Components as Potential Biomarkers for Predicting Disease Progression and Treatment Response
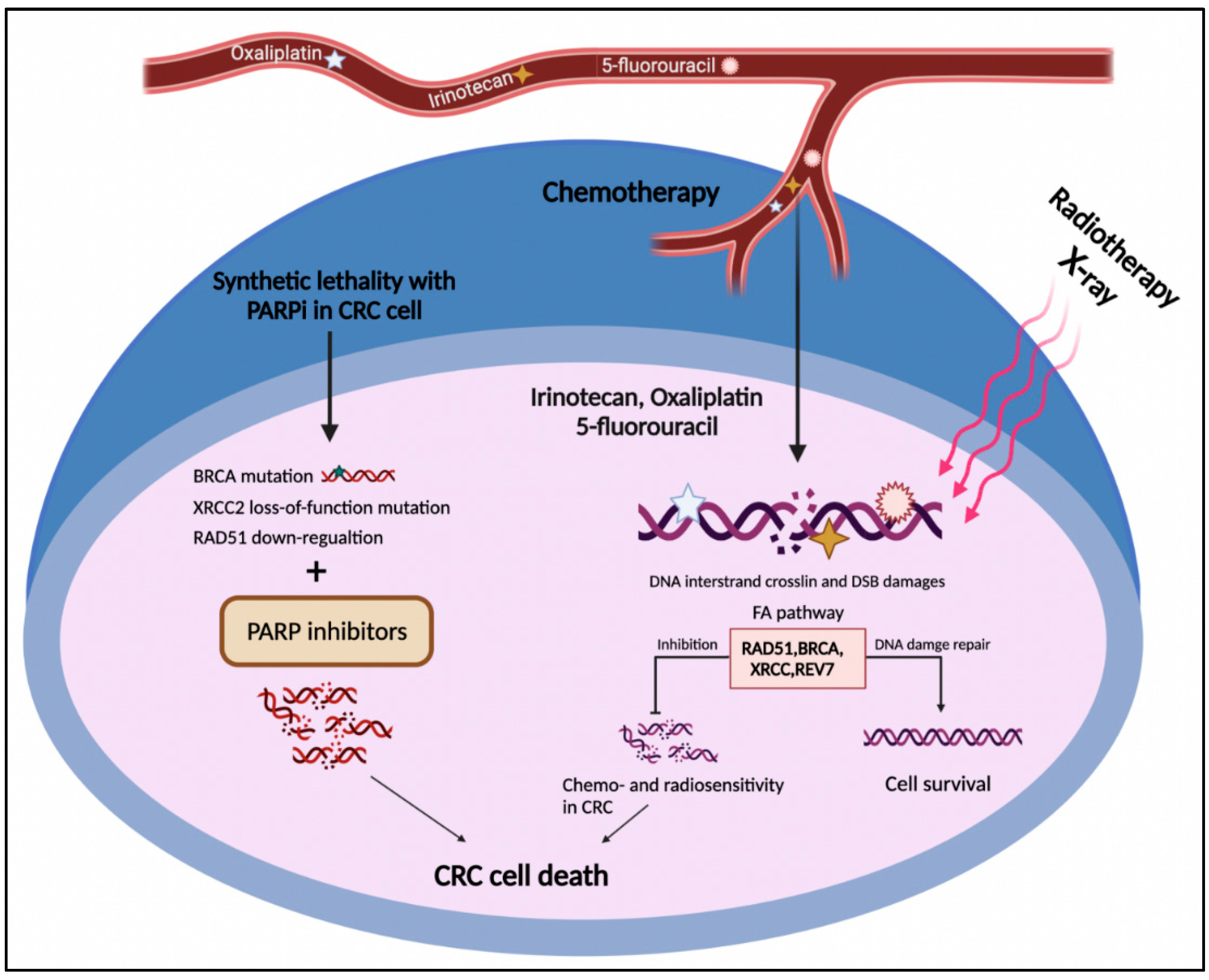
5. FA Components as Promising Therapeutic Targets in CRC
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| APC | APC regulator of WNT signaling pathway |
| ATR | ataxia telangiectasia and RAd3-related kinase |
| BCDX2 complex | RAD51 paralogs (RAD51B, RAD51C, RAD51D) and XRCC2 |
| BMPR1A | bone morphogenetic protein receptor type 1A |
| BRAF | B-Raf proto-oncogene, serine/threonine kinase |
| BRCA | BRCA1 DNA repair associated (alias FANCS) |
| BRCA2 | BRCA2 DNA repair associated (alias FANCD, FANCD1) |
| BRIP1 | BRCA1 interacting helicase 1 (alias FANCJ) |
| CHK1 | checkpoint kinase1 |
| CNV | copy number variation |
| CRC | colorectal cancer |
| dNTP | deoxyribonucleoside triphosphate |
| DSB | double-strand break |
| EOCRC | early-onset CRC |
| ERCC1 | ERCC excision repair 1, endonuclease catalytic subunit |
| ERCC4 | ERCC excision repair 4, endonuclease catalytic subunit (alias FANCQ) |
| FA | Fanconi anemia |
| FAAP | RNA 2′,3′-cyclic phosphate and 5′-OH ligase |
| FAAP100 | FA core complex associated protein 100 |
| FAAP20 | FA core complex associated protein 20 |
| FAAP24 | FA core complex associated protein 24 |
| FAN1 | FANCD2 and FANCI associated nuclease 1 |
| FANC | FA complementation group |
| FANCA | FA complementation group A |
| FANCB | FA complementation group B |
| FANCC | FA complementation group C |
| FANCD1 | BRCA2 DNA repair associated (official symbol BRCA2) |
| FANCD2 | FA complementation group D2 |
| FANCE | FA complementation group E |
| FANCF | FA complementation group F |
| FANCG | FA complementation group G (alias XRCC9) |
| FANCI | FA complementation group I |
| FANCJ | BRCA1 interacting helicase 1 (official symbol BRIP1) |
| FANCL | FA complementation group L |
| FANCM | FA complementation group M |
| FANCN | partner and localizer of BRCA2 (official symbol PALB2) |
| FANCO | RAD51 paralog C (official symbol RAD51C) |
| FANCP | SLX4 structure-specific endonuclease subunit |
| FANCQ | ERCC excision repair 4, endonuclease catalytic subunit (official symbol ERCC4) |
| FANCR | RAD51 recombinase (official symbol RAD51) |
| FANCS | BRCA1 DNA repair associated (official symbol BRCA) |
| FANCT | FA complementation group T/Ubiquitin Conjugating Enzyme E2 T (official symbol UBE2T) |
| FANCU | X-ray repair cross complementing 2 (official symbol XRCC2) |
| FANCV | mitotic arrest deficient 2 like 2 (official symbol: MAD2L2; alias REV7) |
| FANCW | ring finger and WD repeat domain 3 (official symbol RFWD3) |
| FCCTX | familial CRC type X |
| FOLFIRI | fluorouracil/leucovorin, irinotecan |
| FOLFOX | fluorouracil/leucovorin, oxaliplatin |
| G4 | G-quadruplex |
| GEO | Gene Expression Omnibus |
| HR | homologous recombination |
| HRR | homologous recombinational repair |
| ICL | inter-strand crosslink |
| ID2 complex | FANCI-FANCD2 |
| KRAS | Kirsten RAS proto-oncogene |
| LARC | locally advanced rectal cancer |
| mCRC | metastatic CRC |
| MEK1 | mitogen-activated protein kinase kinase 1 (official symbol MAP2K1) |
| MEK2 | mitogen-activated protein kinase kinase 2 (official symbol MAP2K2) |
| MHF1 | centromere protein S (official symbol CENPS) |
| MHF2 | centromere protein X (official symbol CENPX) |
| MLH1 | mutL homolog 1 |
| MLH2 | PMS1 homolog 1, mismatch repair system component (official symbol PMS1) |
| MMR | mismatch repair, |
| MRN complex | MRE11-RAD50-NBS proteins |
| MSH2 | mutS homolog 2 |
| MSH6 | mutS homolog 6 |
| MSI | microsatellite instability |
| MSS | microsatellite stability |
| MUTYH | mutY DNA glycosylase |
| NCI GDC | National Cancer Institute-Genomic Data Commons |
| NER | nucleotide excision repair |
| NGS | next-generation sequencing; |
| NHEJ | non-homologous end joining |
| NTHL | nth like DNA glycosylase |
| PALB2 | partner and localizer of BRCA2 (alias FANCN) |
| PARP1 | poly (ADP-ribose) polymerase 1 |
| PARPi | PARP inhibitor |
| PFS | progression-free survival |
| PMS2 | PMS1 homolog 2, mismatch repair system component |
| POLD1 | DNA polymerase delta 1, catalytic subunit |
| POLE | DNA polymerase epsilon, catalytic subunit |
| POLQ | DNA polymerase theta |
| PPAP | polymerase proofreading–associated polyposis |
| PPI | protein–protein interaction |
| PTEN | phosphatase and tensin homolog |
| PV | pathogenic variation |
| RAD51 | RAD51 recombinase (alias FANCR) |
| RAD51B | RAD51 paralog B |
| RAD51C | RAD51 paralog C (alias FANCO) |
| RAD51D | RAD51 paralog D |
| REV1 | REV1 DNA directed polymerase |
| REV7 | mitotic arrest deficient 2 like 2 (official symbol: MAD2L2; alias FANCV) |
| RFWD3 | ring finger and WD repeat domain 3 (alias FANCW) |
| RNaseH | ribonuclease H |
| SMAD4 | SMAD family member 4 |
| ssDNA | single-strand DNA |
| STK11 | serine/threonine kinase 11 |
| TLS | translesion DNA synthesis |
| TMEJ | polymerase theta-mediated end joining |
| TMS | telomestatin |
| TNM | tumor (lymph) node metastasis staging |
| TP53 | tumor protein p53 |
| UBE2T | Ubiquitin Conjugating Enzyme E2 T (alias FANCT) |
| VEGF | Vascular endothelial growth factor |
| WES | whole-exome sequencing |
| XRCC2 | X-ray repair cross complementing 2 (alias FANCU) |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Rustgi, A.K. The genetics of hereditary colon cancer. Genes Dev. 2007, 21, 2525–2538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, J.M.; Gupta, S.; Burke, C.A.; Axell, L.; Chen, L.-M.; Chung, D.C.; Clayback, K.M.; Dallas, S.; Felder, S.; Gbolahan, O.; et al. NCCN Guidelines® Insights: Genetic/Familial High-Risk Assessment: Colorectal.; Version 1.2021: Featured Updates to the NCCN Guidelines. J. Natl. Compr. Cancer Netw. 2021, 19, 1122–1132. [Google Scholar] [CrossRef]
- Weren, R.D.; Ligtenberg, M.J.; Kets, C.M.; De Voer, R.M.; Verwiel, E.T.; Spruijt, L.; van Zelst-Stams, W.A.; Jongmans, M.C.; Gilissen, C.; Hehir-Kwa, J.Y.; et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat. Genet. 2015, 47, 668–671. [Google Scholar] [CrossRef] [PubMed]
- Valle, L.; de Voer, R.M.; Goldberg, Y.; Sjursen, W.; Försti, A.; Ruiz-Ponte, C.; Caldés, T.; Garré, P.; Olsen, M.F.; Nordling, M.; et al. Update on genetic predisposition to colorectal cancer and polyposis. Mol. Asp. Med. 2019, 69, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Esteban-Jurado, C.; Giménez-Zaragoza, D.; Muñoz, J.; Franch-Expósito, S.; Álvarez-Barona, M.; Ocaña, T.; Cuatrecasas, M.; Carballal, S.; López-Cerón, M.; Marti-Solano, M.; et al. POLE and POLD1 screening in 155 patients with multiple polyps and early-onset colorectal cancer. Oncotarget 2017, 8, 26732–26743. [Google Scholar] [CrossRef] [Green Version]
- Church, J.M. Polymerase Proofreading-Associated Polyposis: A new, dominantly inherited syndrome of hereditary colorectal cancer predisposition. Dis. Colon Rectum 2014, 57, 396–397. [Google Scholar] [CrossRef]
- Magrin, L.; Fanale, D.; Brando, C.; Fiorino, A.; Corsini, L.R.; Sciacchitano, R.; Filorizzo, C.; Dimino, A.; Russo, A.; Bazan, V. POLE, POLD1, and NTHL1: The last but not the least hereditary cancer-predisposing genes. Oncogene 2021, 40, 5893–5901. [Google Scholar] [CrossRef]
- Mur, P.; García-Mulero, S.; del Valle, J.; Magraner-Pardo, L.; Vidal, A.; Pineda, M.; Cinnirella, G.; Martín-Ramos, E.; Pons, T.; López-Doriga, A.; et al. Role of POLE and POLD1 in familial cancer. Genet. Med. 2020, 22, 2089–2100. [Google Scholar] [CrossRef]
- Mankaney, G.; Rouphael, C.; Burke, C.A. Serrated Polyposis Syndrome. Clin. Gastroenterol. Hepatol. 2020, 18, 777–779. [Google Scholar] [CrossRef]
- Velázquez, C.; Lastra, E.; Cobos, F.A.; Abella, L.; De La Cruz, V.; Hernando, B.A.; Hernández, L.; Martínez, N.; Infante, M.; Durán, M. A comprehensive custom panel evaluation for routine hereditary cancer testing: Improving the yield of germline mutation detection. J. Transl. Med. 2020, 18, 232. [Google Scholar] [CrossRef] [PubMed]
- Boussios, S.; Ozturk, M.A.; Moschetta, M.; Karathanasi, A.; Zakynthinakis-Kyriakou, N.; Katsanos, K.H.; Christodoulou, D.K.; Pavlidis, N. The Developing Story of Predictive Biomarkers in Colorectal Cancer. J. Pers. Med. 2019, 9, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mini, E.; Landini, I.; Di Paolo, A.; Ravegnini, G.; Saponara, S.; Frosini, M.; Lapucci, A.; Nobili, S. Predictive ‘Omic’ biomarkers of drug response: Colorectal cancer as a model. In Anti-Angiogenic Drugs as Chemosensitizers in Cancer Therapy; Cancer Sensitizing Agents for Chemotherapy Series; Morbidelli, L., Ed.; Academic Press (Elsevier): London, UK, 2022; Volume 18, pp. 199–240. [Google Scholar]
- Liu, W.; Palovcak, A.; Li, F.; Zafar, A.; Yuan, F.; Zhang, Y. Fanconi anemia pathway as a prospective target for cancer intervention. Cell Biosci. 2020, 10, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolinjivadi, A.M.; Crismani, W.; Ngeow, J. Emerging functions of Fanconi anemia genes in replication fork protection pathways. Hum. Mol. Genet. 2020, 29, R158–R164. [Google Scholar] [CrossRef]
- Woodward, E.R.; Meyer, S. Fanconi Anaemia, Childhood Cancer and the BRCA Genes. Genes 2021, 12, 1520. [Google Scholar] [CrossRef]
- Nalepa, G.; Clapp, D.W. Fanconi anaemia and cancer: An intricate relationship. Nat. Rev. Cancer 2018, 18, 168–185. [Google Scholar] [CrossRef]
- Nepal, M.; Che, R.; Zhang, J.; Ma, C.; Fei, P. Fanconi anemia Signaling and Cancer. Trends Cancer 2017, 3, 840–856. [Google Scholar] [CrossRef]
- Michl, J.; Zimmer, J.; Tarsounas, M. Interplay between Fanconi anemia and homologous recombination pathways in genome integrity. EMBO J. 2016, 35, 909–923. [Google Scholar] [CrossRef]
- Howlett, N.G.; Taniguchi, T.; Olson, S.; Cox, B.; Waisfisz, Q.; De Die-Smulders, C.; Persky, N.; Grompe, M.; Joenje, H.; Pals, G.; et al. Biallelic Inactivation of BRCA2 in Fanconi anemia. Science 2002, 297, 606–609. [Google Scholar] [CrossRef]
- Milletti, G.; Strocchio, L.; Pagliara, D.; Girardi, K.; Carta, R.; Mastronuzzi, A.; Locatelli, F.; Nazio, F. Canonical and Noncanonical Roles of Fanconi anemia Proteins: Implications in Cancer Predisposition. Cancers 2020, 12, 2684. [Google Scholar] [CrossRef] [PubMed]
- Risinger, M.A.; Groden, J. Crosslinks and crosstalk: Human cancer syndromes and DNA repair defects. Cancer Cell 2004, 6, 539–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, C.-B.; Wu, H.-T.; Zhang, M.-L.; Liu, J.; Zhang, G.-J. Fanconi anemia Pathway: Mechanisms of Breast Cancer Predisposition Development and Potential Therapeutic Targets. Front. Cell Dev. Biol. 2020, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.H.; Ni, Y.; Li, S.-T.; Teo, J.X.; Ishak, N.D.B.; Lim, W.K.; Ngeow, J. Spectrum of Germline Mutations within Fanconi anemia–Associated Genes Across Populations of Varying Ancestry. JNCI Cancer Spectr. 2021, 5, pkaa117. [Google Scholar] [CrossRef]
- Niraj, J.; Färkkilä, A.; D’Andrea, A.D. The Fanconi anemia Pathway in Cancer. Annu. Rev. Cancer Biol. 2019, 3, 457–478. [Google Scholar] [CrossRef]
- Boussios, S.; Mikropoulos, C.; Samartzis, E.; Karihtala, P.; Moschetta, M.; Sheriff, M.; Karathanasi, A.; Sadauskaite, A.; Rassy, E.; Pavlidis, N. Wise Management of Ovarian Cancer: On the Cutting Edge. J. Pers. Med. 2020, 10, 41. [Google Scholar] [CrossRef]
- Yurgelun, M.B.; Kulke, M.H.; Fuchs, C.S.; Allen, B.A.; Uno, H.; Hornick, J.; Ukaegbu, C.I.; Brais, L.K.; McNamara, P.G.; Mayer, R.J.; et al. Cancer Susceptibility Gene Mutations in Individuals with Colorectal Cancer. J. Clin. Oncol. 2017, 35, 1086–1095. [Google Scholar] [CrossRef]
- Esteban-Jurado, C.; Franch-Expósito, S.; Muñoz, J.; Ocaña, T.; Carballal, S.; López-Cerón, M.; Cuatrecasas, M.; Vila-Casadesús, M.; Lozano, J.J.; Serra, E.; et al. The Fanconi anemia DNA damage repair pathway in the spotlight for germline predisposition to colorectal cancer. Eur. J. Hum. Genet. 2016, 24, 1501–1505. [Google Scholar] [CrossRef]
- Gong, R.; He, Y.; Liu, X.-Y.; Wang, H.-Y.; Sun, L.-Y.; Yang, X.-H.; Li, B.; Cao, X.-K.; Ye, Z.-L.; Kong, L.-H.; et al. Mutation spectrum of germline cancer susceptibility genes among unselected Chinese colorectal cancer patients. Cancer Manag. Res. 2019, 11, 3721–3739. [Google Scholar] [CrossRef] [Green Version]
- Pearlman, R.; Frankel, W.L.; Swanson, B.; Zhao, W.; Yilmaz, A.; Miller, K.; Bacher, J.; Bigley, C.; Nelsen, L.; Goodfellow, P.J.; et al. Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations among Patients with Early-Onset Colorectal Cancer. JAMA Oncol. 2017, 3, 464–471. [Google Scholar] [CrossRef]
- Bertelsen, B.; Tuxen, I.V.; Yde, C.W.; Gabrielaite, M.; Torp, M.H.; Kinalis, S.; Oestrup, O.; Rohrberg, K.; Spangaard, I.; Santoni-Rugiu, E.; et al. High frequency of pathogenic germline variants within homologous recombination repair in patients with advanced cancer. NPJ Genom. Med. 2019, 4, 13. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; D’Andrea, A. Fanconi anemia pathway. Curr. Biol. 2017, 27, R986–R988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Li, L. DNA crosslinking damage and cancer—A tale of friend and foe. Transl. Cancer Res. 2013, 2, 144–154. [Google Scholar] [CrossRef]
- Zhang, J.; Dewar, J.M.; Budzowska, M.; Motnenko, A.; Cohn, M.A.; Walter, J.C. DNA interstrand cross-link repair requires replication-fork convergence. Nat. Struct. Mol. Biol. 2015, 22, 242–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharjee, S.; Nandi, S. DNA damage response and cancer therapeutics through the lens of the Fanconi anemia DNA repair pathway. Cell Commun. Signal. 2017, 15, 41. [Google Scholar] [CrossRef] [Green Version]
- Helbling-Leclerc, A.; Garcin, C.; Rosselli, F. Beyond DNA repair and chromosome instability—Fanconi anaemia as a cellular senescence-associated syndrome. Cell Death Differ. 2021, 28, 1159–1173. [Google Scholar] [CrossRef]
- Clauson, C.; Schärer, O.D.; Niedernhofer, L. Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012732. [Google Scholar] [CrossRef]
- Roy, U.; Schärer, O.D. Involvement of translesion synthesis DNA polymerases in DNA interstrand crosslink repair. DNA Repair 2016, 44, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Haince, J.-F.; McDonald, D.; Rodrigue, A.; Déry, U.; Masson, J.-Y.; Hendzel, M.J.; Poirier, G.G. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008, 283, 1197–1208. [Google Scholar] [CrossRef] [Green Version]
- Reilly, N.M.; Novara, L.; Di Nicolantonio, F.; Bardelli, A. Exploiting DNA repair defects in colorectal cancer. Mol. Oncol. 2019, 13, 681–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurini, E.; Marson, D.; Fermeglia, A.; Aulic, S.; Fermeglia, M.; Pricl, S. Role of Rad51 and DNA repair in cancer: A molecular perspective. Pharmacol. Ther. 2020, 208, 107492. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Chapman, J.R.; Brandsma, I.; Yuan, J.; Mistrik, M.; Bouwman, P.; Bartkova, J.; Gogola, E.; Warmerdam, D.; Barazas, M.; et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 2015, 521, 541–544. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Zhang, Y.; Zhang, J.; Qi, C.; Liu, D.; Wang, Z.; Li, Y.; Ji, C.; Li, J.; Lin, X.; et al. Germline Profiling and Molecular Characterization of Early Onset Metastatic Colorectal Cancer. Front. Oncol. 2020, 10, 568911. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, H.; Iwatsuki, M.; Mimori, K.; Sato, T.; Johansson, F.; Toh, H.; Watanabe, M.; Mori, M. FANCD2 mRNA overexpression is a bona fide indicator of lymph node metastasis in human colorectal cancer. Ann. Surg. Oncol. 2010, 17, 2341–2348. [Google Scholar] [CrossRef]
- Landais, I.; Hiddingh, S.; McCarroll, M.; Yang, C.; Sun, A.; Turker, M.S.; Snyder, J.P.; Hoatlin, M.E. Monoketone analogs of curcumin, a new class of Fanconi anemia pathway inhibitors. Mol. Cancer 2009, 8, 133. [Google Scholar] [CrossRef] [Green Version]
- Her, J.; Ray, C.; Altshuler, J.; Zheng, H.; Bunting, S.F. 53BP1 Mediates ATR-Chk1 Signaling and Protects Replication Forks under Conditions of Replication Stress. Mol. Cell Biol. 2018, 38, e00472-17. [Google Scholar] [CrossRef] [Green Version]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Saxena, S.; Dixit, S.; Somyajit, K.; Nagaraju, G. ATR Signaling Uncouples the Role of RAD51 Paralogs in Homologous Recombination and Replication Stress Response. Cell Rep. 2019, 29, 551–559.e4. [Google Scholar] [CrossRef] [Green Version]
- Liao, H.; Ji, F.; Helleday, T.; Ying, S. Mechanisms for stalled replication fork stabilization: New targets for synthetic lethality strategies in cancer treatments. EMBO Rep. 2018, 19, e46263. [Google Scholar] [CrossRef]
- Skourti-Stathaki, K.; Proudfoot, N.J. A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 2014, 28, 1384–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Rubio, M.L.; Pérez-Calero, C.; Barroso, S.I.; Tumini, E.; Moyano, E.H.; Rosado, I.V.; Aguilera, A. The Fanconi anemia Pathway Protects Genome Integrity from R-loops. PLoS Genet. 2015, 11, e1005674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palovcak, A.; Liu, W.; Yuan, F.; Zhang, Y. Maintenance of genome stability by Fanconi anemia proteins. Cell Biosci. 2017, 7, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kais, Z.; Rondinelli, B.; Holmes, A.; O’Leary, C.; Kozono, D.; D’Andrea, A.D.; Ceccaldi, R. FANCD2 Maintains Fork Stability in BRCA1/2-Deficient Tumors and Promotes Alternative End-Joining DNA Repair. Cell Rep. 2016, 15, 2488–2499. [Google Scholar] [CrossRef] [Green Version]
- Schwab, R.A.; Nieminuszczy, J.; Shah, F.; Langton, J.; Martinez, D.L.; Liang, C.-C.; Cohn, M.A.; Gibbons, R.J.; Deans, A.J.; Niedzwiedz, W. The Fanconi anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Mol. Cell 2015, 60, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Maffia, A.; Ranise, C.; Sabbioneda, S. From R-Loops to G-Quadruplexes: Emerging New Threats for the Replication Fork. Int. J. Mol. Sci. 2020, 21, 1506. [Google Scholar] [CrossRef] [Green Version]
- Teng, F.-Y.; Jiang, Z.-Z.; Guo, M.; Tan, X.-Z.; Chen, F.; Xi, X.-G.; Xu, Y. G-quadruplex DNA: A novel target for drug design. Cell Mol. Life Sci. 2021, 78, 6557–6583. [Google Scholar] [CrossRef]
- Biffi, G.; Tannahill, D.; Miller, J.; Howat, W.J.; Balasubramanian, S. Elevated levels of G-quadruplex formation in human stomach and liver cancer tissues. PLoS ONE 2014, 9, e102711. [Google Scholar] [CrossRef]
- Lowran, K.; Campbell, L.; Popp, P.; Wu, C.G. Assembly of a G-Quadruplex Repair Complex by the FANCJ DNA Helicase and the REV1 Polymerase. Genes 2019, 11, 5. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.G.; Spies, M. G-quadruplex recognition and remodeling by the FANCJ helicase. Nucleic Acids Res. 2016, 44, 8742–8753. [Google Scholar] [CrossRef] [Green Version]
- Awadasseid, A.; Ma, X.; Wu, Y.; Zhang, W. G-quadruplex stabilization via small-molecules as a potential anti-cancer strategy. Biomed. Pharmacother. 2021, 139, 111550. [Google Scholar] [CrossRef] [PubMed]
- Francisco, A.P.; Paulo, A. Oncogene Expression Modulation in Cancer Cell Lines by DNA G-Quadruplex-Interactive Small Molecules. Curr. Med. Chem. 2018, 24, 4873–4904. [Google Scholar] [CrossRef] [PubMed]
- Estep, K.N.; Butler, T.J.; Ding, J.; Brosh, R.M. G4-Interacting DNA Helicases and Polymerases: Potential Therapeutic Targets. Curr. Med. Chem. 2019, 26, 2881–2897. [Google Scholar] [CrossRef] [PubMed]
- Brito, H.; Martins, A.C.; Lavrado, J.; Mendes, M.; Francisco, A.P.; Santos, S.A.; Ohnmacht, S.A.; Kim, N.-S.; Rodrigues, C.M.; Moreira, R.; et al. Targeting KRAS Oncogene in Colon Cancer Cells with 7-Carboxylate Indolo[3,2-b]quinoline Tri-Alkylamine Derivatives. PLoS ONE 2015, 10, e0126891. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Shin-Ya, K.; Brosh, R.M., Jr. FANCJ helicase defective in Fanconia anemia and breast cancer unwinds G-quadruplex DNA to defend genomic stability. Mol. Cell Biol. 2008, 28, 4116–4128. [Google Scholar] [CrossRef] [Green Version]
- Henderson, A.; Wu, Y.; Huang, Y.C.; Chavez, E.A.; Platt, J.; Johnson, F.B.; Brosh, R.M., Jr.; Sen, D.; Lansdorp, P.M. Detection of G-quadruplex DNA in mammalian cells. Nucleic Acids Res. 2014, 42, 860–869. [Google Scholar] [CrossRef] [Green Version]
- Kolinjivadi, A.M.; Sannino, V.; De Antoni, A.; Técher, H.; Baldi, G.; Costanzo, V. Moonlighting at replication forks—A new life for homologous recombination proteins BRCA1, BRCA2 and RAD51. FEBS Lett. 2017, 591, 1083–1100. [Google Scholar] [CrossRef]
- García-De-Teresa, B.; Rodríguez, A.; Frias, S. Chromosome Instability in Fanconi anemia: From Breaks to Phenotypic Consequences. Genes 2020, 11, 1528. [Google Scholar] [CrossRef]
- Chen, X.; Bosques, L.; Sung, P.; Kupfer, G.M. A novel role for non-ubiquitinated FANCD2 in response to hydroxyurea-induced DNA damage. Oncogene 2016, 35, 22–34. [Google Scholar] [CrossRef]
- Ghose, A.; Moschetta, M.; Pappas-Gogos, G.; Sheriff, M.; Boussios, S. Genetic Aberrations of DNA Repair Pathways in Prostate Cancer: Translation to the Clinic. Int. J. Mol. Sci. 2021, 22, 9783. [Google Scholar] [CrossRef]
- Chang, P.-Y.; Chang, S.-C.; Wang, M.-C.; Chen, J.-S.; Tsai, W.-S.; You, J.-F.; Chen, C.-C.; Liu, H.-L.; Chiang, J.-M. Pathogenic Germline Mutations of DNA Repair Pathway Components in Early-Onset Sporadic Colorectal Polyp and Cancer Patients. Cancers 2020, 12, 3560. [Google Scholar] [CrossRef] [PubMed]
- AlDubayan, S.H.; Giannakis, M.; Moore, N.D.; Han, G.C.; Reardon, B.; Hamada, T.; Mu, X.J.; Nishihara, R.; Qian, Z.; Liu, L.; et al. Inherited DNA-Repair Defects in Colorectal Cancer. Am. J. Hum. Genet. 2018, 102, 401–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degrolard-Courcet, E.; Sokolowska, J.; Padeano, M.-M.; Guiu, S.; Bronner, M.; Chery, C.; Coron, F.; Lepage, C.; Chapusot, C.; Loustalot, C.; et al. Development of primary early-onset colorectal cancers due to biallelic mutations of the FANCD1/BRCA2 gene. Eur. J. Hum. Genet. 2014, 22, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Palmieri Molecular analysis of Fanconi anemia and mismatch repair genes in patients with colorectal carcinoma. Oncol. Rep. 2011, 25, 899–904. [CrossRef] [Green Version]
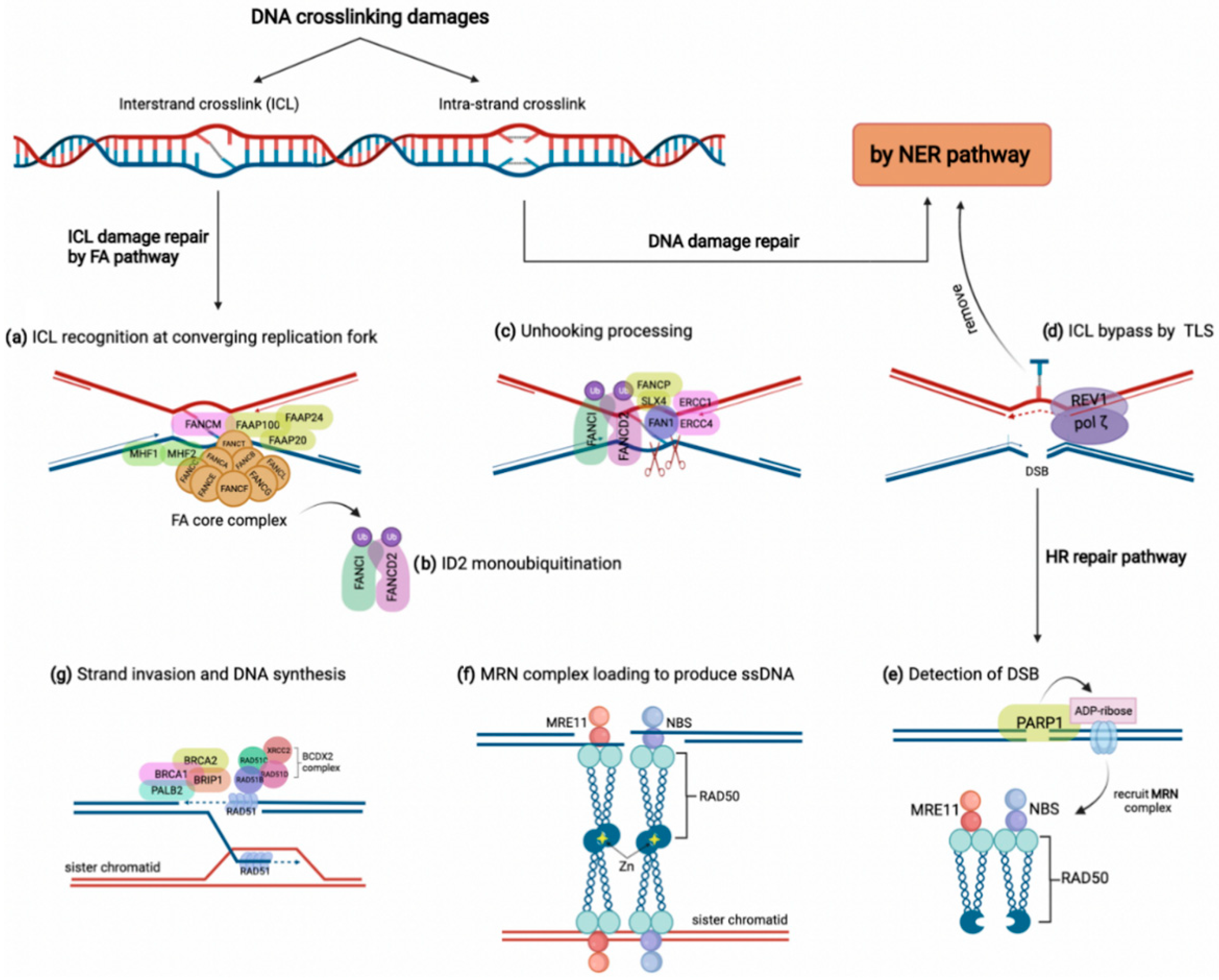
- Tham, K.-C.; Kanaar, R.; Lebbink, J.H.G. Mismatch repair and homeologous recombination. DNA Repair 2016, 38, 75–83. [Google Scholar] [CrossRef]
- Rahman, N.; Scott, R.H. Cancer genes associated with phenotypes in monoallelic and biallelic mutation carriers: New lessons from old players. Hum. Mol. Genet. 2007, 16, R60–R66. [Google Scholar] [CrossRef] [Green Version]
- Peng, M.; Litman, R.; Xie, J.; Sharma, S.; Brosh, R.M., Jr.; Cantor, S.B. The FANCJ/MutLα interaction is required for correction of the cross-link response in FA-J cells. EMBO J. 2007, 26, 3238–3249. [Google Scholar] [CrossRef]
- Wang, Y.; Cortez, D.; Yazdi, P.; Neff, N.; Elledge, S.J.; Qin, J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000, 14, 927–939. [Google Scholar] [CrossRef]
- Williams, S.A.; Wilson, J.B.; Clark, A.P.; Mitson-Salazar, A.; Tomashevski, A.; Ananth, S.; Glazer, P.M.; Semmes, O.J.; Bale, A.E.; Jones, N.J.; et al. Functional and physical interaction between the mismatch repair and FA-BRCA pathways. Hum. Mol. Genet. 2011, 20, 4395–4410. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Heald, B.; Kupfer, S.S.; Hampel, H.; Church, J.; Dudley, B.; Hall, M.J.; Mork, M.E.; Singh, A.; Stoffel, E.; Stoll, J.; et al. Collaborative Group of the Americas on Inherited Gastrointestinal Cancer Position statement on multigene panel testing for patients with colorectal cancer and/or polyposis. Fam. Cancer 2020, 19, 223–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akcay, I.M.; Celik, E.; Agaoglu, N.B.; Alkurt, G.; Msc, T.K.A.; Yildiz, J.; Enc, F.; Kir, G.; Canbek, S.; Kilic, A.; et al. Germline pathogenic variant spectrum in 25 cancer susceptibility genes in Turkish breast and colorectal cancer patients and elderly controls. Int. J. Cancer 2021, 148, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Toh, M.R.; Chiang, J.B.; Chong, S.T.; Chan, S.H.; Ishak, N.D.B.; Courtney, E.; Lee, W.H.; Syed Abdillah Al, S.M.F.B.; Allen, J.C.; Lim, K.H.; et al. Germline Pathogenic Variants in Homologous Recombination and DNA Repair Genes in an Asian Cohort of Young-Onset Colorectal Cancer. JNCI Cancer Spectr. 2018, 2, pky054. [Google Scholar] [CrossRef]
- Garre, P.; Martín, L.; Sanz, J.; Romero, A.; Tosar, A.; Bando, I.; Llovet, P.; Diaque, P.; García-Paredes, B.; Rubio, P.G.; et al. BRCA2gene: A candidate for clinical testing in familial colorectal cancer type X. Clin. Genet. 2015, 87, 582–587. [Google Scholar] [CrossRef]
- Yurgelun, M.B.; Allen, B.; Kaldate, R.R.; Bowles, K.R.; Judkins, T.; Kaushik, P.; Roa, B.B.; Wenstrup, R.J.; Hartman, A.-R.; Syngal, S. Identification of a Variety of Mutations in Cancer Predisposition Genes in Patients with Suspected Lynch Syndrome. Gastroenterology 2015, 149, 604–613.e20. [Google Scholar] [CrossRef] [Green Version]
- Stoffel, E.M.; Koeppe, E.; Everett, J.; Ulintz, P.; Kiel, M.; Osborne, J.; Williams, L.; Hanson, K.; Gruber, S.B.; Rozek, L.S. Germline Genetic Features of Young Individuals with Colorectal Cancer. Gastroenterology 2018, 154, 897–905.e1. [Google Scholar] [CrossRef]
- Naseem, M.; Xiu, J.; Salem, M.E.; Goldberg, R.M.; Vanderwalde, A.M.; Grothey, A.; Philip, P.A.; Seeber, A.; Puccini, A.; Tokunaga, R.; et al. Characteristics of colorectal cancer (CRC) patients with BRCA1 and BRCA2 mutations. J. Clin. Oncol. 2019, 37, 606. [Google Scholar] [CrossRef] [Green Version]
- Mikaeel, R.R.; Young, J.P.; Li, Y.; Smith, E.; Horsnell, M.; Uylaki, W.; Rico, G.T.; Poplawski, N.K.; Hardingham, J.E.; Tomita, Y.; et al. Survey of germline variants in cancer-associated genes in young adults with colorectal cancer. Genes Chromosom. Cancer 2021, 65, 105–113. [Google Scholar] [CrossRef]
- Suchy, J.; Cybulski, C.; Górski, B.; Huzarski, T.; Byrski, T.; Dębniak, T.; Gronwald, J.; Jakubowska, A.; Wokołorczyk, D.; Kurzawski, G.; et al. BRCA1 mutations and colorectal cancer in Poland. Fam. Cancer 2010, 9, 541–544. [Google Scholar] [CrossRef]
- Phelan, C.M.; Iqbal, J.; Lynch, H.T.; Lubinski, J.; Gronwald, J.; Moller, P.; Ghadirian, P.; Foulkes, W.D.; Armel, S.; Eisen, A.; et al. Incidence of colorectal cancer in BRCA1 and BRCA2 mutation carriers: Results from a follow-up study. Br. J. Cancer 2014, 110, 530–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullinane, C.M.; Creavin, B.; O’Connell, E.P.; Kelly, L.; O’Sullivan, M.J.; Corrigan, M.A.; Redmond, H.P. Risk of colorectal cancer associated with BRCA1 and/or BRCA2 mutation carriers: Systematic review and meta-analysis. Br. J. Surg. 2020, 107, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Maccaroni, E.; Giampieri, R.; Lenci, E.; Scortichini, L.; Bianchi, F.; Belvederesi, L.; Brugiati, C.; Pagliaretta, S.; Ambrosini, E.; Berardi, R. BRCA mutations and gastrointestinal cancers: When to expect the unexpected? World J. Clin. Oncol. 2021, 12, 565–580. [Google Scholar] [CrossRef] [PubMed]
- Martín-Morales, L.; Garre, P.; Lorca, V.; Cazorla, M.; Llovet, P.; Bando, I.; García-Barberan, V.; González-Morales, M.L.; Esteban-Jurado, C.; De La Hoya, M.; et al. BRIP1, a Gene Potentially Implicated in Familial Colorectal Cancer Type X. Cancer Prev. Res. 2020, 14, 185–194. [Google Scholar] [CrossRef]
- Ali, M.; DeLozier, C.D.; Chaudhary, U. BRIP-1 germline mutation and its role in colon cancer: Presentation of two case reports and review of literature. BMC Med. Genet. 2019, 20, 75. [Google Scholar] [CrossRef] [Green Version]
- Cannon-Albright, L.A.; Teerlink, C.C.; Stevens, J.; Snow, A.K.; Thompson, B.A.; Bell, R.; Nguyen, K.N.; Sargent, N.R.; Kohlmann, W.K.; Neklason, D.W.; et al. FANCM c5791C>T stopgain mutation (rs144567652) is a familial colorectal cancer risk factor. Mol. Genet. Genom. Med. 2020, 8, e1532. [Google Scholar] [CrossRef]
- Innocenti, F.; Rashid, N.; Wancen, M.; Ou, F.-S.; Qu, X.; Denning, S.; Bertagnoli, M.; Blanke, C.D.; Venook, A.; Kabbarah, O.; et al. Next-generation sequencing (NGS) in metastatic colorectal cancer (mCRC): Novel mutated genes and their effect on response to therapy (Alliance). Ann. Oncol. 2019, 30, 198–199. [Google Scholar] [CrossRef]
- Martínez-Jiménez, F.; Muiños, F.; Sentís, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef]
- GDC Data Portal—National Cancer Institute. Available online: www.portal.gdc.cancer.gov (accessed on 26 February 2022).
- Qin, C.-J.; Song, X.-M.; Chen, Z.-H.; Ren, X.-Q.; Xu, K.-W.; Jing, H.; He, Y.-L. XRCC2 as a predictive biomarker for radioresistance in locally advanced rectal cancer patients undergoing preoperative radiotherapy. Oncotarget 2015, 6, 32193–32204. [Google Scholar] [CrossRef]
- Marsh, S.; McLeod, H.; Dolan, E.; Rabik, C.; Shukla, S.J.; Gong, L.; Hernandez-Boussard, T.; Lou, X.J.; Klein, T.E.; Altman, R.B. Platinum pathway. Pharmacogenet. Genom. 2009, 19, 563–564. [Google Scholar] [CrossRef] [Green Version]
- Cepeda, V.; Fuertes, M.A.; Castilla, J.; Alonso, C.; Quevedo, C.; Perez, J.M. Biochemical Mechanisms of Cisplatin Cytotoxicity. Anti-Cancer Agents Med. Chem. 2007, 7, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, R.; Kitao, H.; Fujinaka, Y.; Yamashita, N.; Iimori, M.; Tokunaga, E.; Yamashita, N.; Morita, M.; Kakeji, Y.; Maehara, Y. FANCJ expression predicts the response to 5-fluorouracil-based chemotherapy in MLH1-proficient colorectal cancer. Ann. Surg. Oncol. 2012, 19, 3627–3635. [Google Scholar] [CrossRef] [PubMed]
- Ihara, K.; Yamaguchi, S.; Ueno, N.; Tani, Y.; Shida, Y.; Ogata, H.; Domeki, Y.; Okamoto, K.; Nakajima, M.; Sasaki, K.; et al. Expression of DNA double-strand break repair proteins predicts the response and prognosis of colorectal cancer patients undergoing oxaliplatin-based chemotherapy. Oncol. Rep. 2016, 35, 1349–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Li, X.; Huang, W.; Chen, Y.; Wang, B.; Liu, X. Ubiquitin-conjugating enzyme E2T(UBE2T) promotes colorectal cancer progression by facilitating ubiquitination and degradation of p53. Clin. Res. Hepatol. Gastroenterol. 2021, 45, 101493. [Google Scholar] [CrossRef]
- Wu, X.; Liu, G.; Liu, R.; He, J.; Wang, G.; Zhang, H.; Liu, T.; Bai, J.; Cheng, N.; Qiu, J. Expression of ubiquitin-conjugating enzyme E2T in colorectal cancers and clinical implications. Oncol. Lett. 2020, 20, 275. [Google Scholar] [CrossRef]
- Zhang, Y.-Z.; An, J.-H.; Liu, Y.-X.; Wu, X.-C.; Han, S.-S.; Ren, X.-Q.; Qin, C.-J. XRCC2-Deficient Cells are Highly Sensitive to 5-Fluorouracil in Colorectal Cancer. Cell. Physiol. Biochem. 2017, 43, 1207–1219. [Google Scholar] [CrossRef] [Green Version]
- Tennstedt, P.; Fresow, R.; Simon, R.; Marx, A.; Terracciano, L.; Petersen, C.; Sauter, G.; Dikomey, E.; Borgmann, K. RAD51 overexpression is a negative prognostic marker for colorectal adenocarcinoma. Int. J. Cancer 2013, 132, 2118–2126. [Google Scholar] [CrossRef]
- Lemonidis, K.; Arkinson, C.; Rennie, M.L.; Walden, H. Mechanism, specificity, and function of FANCD2-FANCI ubiquitination and deubiquitination. FEBS J. 2021. [Google Scholar] [CrossRef]
- Liu, C.; Gao, J.; LE, Y.; Zhong, X.; Chen, Y.; Wang, H. Expression of FANCD2 in Colorectal Cancer and Its Correlation with Prognosis. Chin. J. Gastroenterol. 2014, 404–407. [Google Scholar]
- Lee, J.-H.; Bae, A.-N.; Jung, S.-J. Clinicopathological and Prognostic Characteristics of RAD51 in Colorectal Cancer. Medicina 2020, 56, 48. [Google Scholar] [CrossRef] [Green Version]
- Slupianek, A.; Hoser, G.; Majsterek, I.; Bronisz, A.; Malecki, M.; Blasiak, J.; Fishel, R.; Skorski, T. Fusion tyrosine kinases induce drug resistance by stimulation of homology-dependent recombination repair, prolongation of G2/M phase, and protection from apoptosis. Mol. Cell. Biol. 2002, 22, 4189–4201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vispe, S.; Cazaux, C.; Lesca, C.; Defais, M. Overexpression of Rad51 protein stimulates homologous recombination and increases resistance of mammalian cells to ionizing radiation. Nucleic Acids Res. 1998, 26, 2859–2864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brosh, R.M., Jr.; Cantor, S.B. Molecular and cellular functions of the FANCJ DNA helicase defective in cancer and in Fanconi anemia. Front. Genet. 2014, 5, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, C.; Peng, Y.; He, Y.; Chen, G.; Chen, H. Low levels of BRCA1 protein expression predict a worse prognosis in stage I–II colon cancer. Int. J. Biol. Markers 2021, 36, 47–53. [Google Scholar] [CrossRef]
- Leaf, S.; Carlsen, L.; El-Deiry, W.S. Opposing effects of BRCA1 mRNA expression on patient survival in breast and colorectal cancer and variations among African American, Asian, and younger patients. Oncotarget 2021, 12, 1992–2005. [Google Scholar] [CrossRef]
- Yuanming, L.; Lineng, Z.; Baorong, S.; Junjie, P.; Sanjun, C. BRCA1 and ERCC1mRNA levels are associated with lymph node metastasis in Chinese patients with colorectal cancer. BMC Cancer 2013, 13, 103. [Google Scholar] [CrossRef] [Green Version]
- Rajarajan, S.; Anupamam, A.; Jose, B.; Correa, M.; Sengupta, S.; Prabhu, J.S. Identification of colorectal cancers with defective DNA damage repair by immunohistochemical profiling of mismatch repair proteins, CDX2 and BRCA1. Mol. Clin. Oncol. 2020, 13, 57. [Google Scholar] [CrossRef]
- Wang, G.-H.; Zhao, C.-M.; Huang, Y.; Wang, W.; Zhang, S.; Wang, X. BRCA1 and BRCA2 expression patterns and prognostic significance in digestive system cancers. Hum. Pathol. 2018, 71, 135–144. [Google Scholar] [CrossRef]
- Grabsch, H.; Dattani, M.; Barker, L.; Maughan, N.; Maude, K.; Hansen, O.; Gabbert, H.E.; Quirke, P.; Mueller, W. Expression of DNA double-strand break repair proteins ATM and BRCA1 predicts survival in colorectal cancer. Clin. Cancer Res. 2006, 12, 1494–1500. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Setton, J.; Lee, N.Y.; Riaz, N.; Powell, S.N. The therapeutic significance of mutational signatures from DNA repair deficiency in cancer. Nat. Commun. 2018, 9, 3292. [Google Scholar] [CrossRef]
- Hosoya, N.; Miyagawa, K. Targeting DNA damage response in cancer therapy. Cancer Sci. 2014, 105, 370–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Tang, C.; Yao, Y.; Chen, X.; Zhou, C.; Wei, Z.; Xing, F.; Chen, L.; Cai, X.; Zhang, Z.; et al. The tumor therapy landscape of synthetic lethality. Nat. Commun. 2021, 12, 1275. [Google Scholar] [CrossRef] [PubMed]
- Tomasini, P.P.; Guecheva, T.N.; Leguisamo, N.M.; Péricart, S.; Brunac, A.-C.; Hoffmann, J.S.; Saffi, J. Analyzing the Opportunities to Target DNA Double-Strand Breaks Repair and Replicative Stress Responses to Improve Therapeutic Index of Colorectal Cancer. Cancers 2021, 13, 3130. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Rachmat, R.; Enyioma, S.; Ghose, A.; Revythis, A.; Boussios, S. BRCA Mutations in Prostate Cancer: Assessment, Implications and Treatment Considerations. Int. J. Mol. Sci. 2021, 22, 12628. [Google Scholar] [CrossRef]
- Krajewska, M.; Fehrmann, R.S.; Schoonen, P.M.; Labib, S.; de Vries, E.G.; Franke, L.; Van Vugt, M.A.T.M. ATR inhibition preferentially targets homologous recombination-deficient tumor cells. Oncogene 2015, 34, 3474–3481. [Google Scholar] [CrossRef]
- Gaillard, H.; Garcia-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Dyczkowski, J.; Beißbarth, T.; Gaedcke, J.; Mansour, W.Y.; Borgmann, K.; Dobbelstein, M. 5-Fluorouracil sensitizes colorectal tumor cells towards double stranded DNA breaks by interfering with homologous recombination repair. Oncotarget 2015, 6, 12574–12586. [Google Scholar] [CrossRef]
- Manic, G.; Musella, M.; Corradi, F.; Sistigu, A.; Vitale, S.; Rehim, S.S.A.; Mattiello, L.; Malacaria, E.; Galassi, C.; Signore, M.; et al. Control of replication stress and mitosis in colorectal cancer stem cells through the interplay of PARP1, MRE11 and RAD51. Cell Death Differ. 2021, 28, 2060–2082. [Google Scholar] [CrossRef]
- Manic, G.; Signore, M.; Sistigu, A.; Russo, G.; Corradi, F.; Siteni, S.; Musella, M.; Vitale, S.; De Angelis, M.L.; Pallocca, M.; et al. CHK1-targeted therapy to deplete DNA replication-stressed, p53-deficient, hyperdiploid colorectal cancer stem cells. Gut 2018, 67, 903–917. [Google Scholar] [CrossRef]
- Mattiello, L.; Rehim, S.S.A.; Musella, M.; Sistigu, A.; Guarracino, A.; Vitale, S.; Corradi, F.; Galassi, C.; Sperati, F.; Manic, G.; et al. The Targeting of MRE11 or RAD51 Sensitizes Colorectal Cancer Stem Cells to CHK1 Inhibition. Cancers 2021, 13, 1957. [Google Scholar] [CrossRef]
- Smeby, J.; Kryeziu, K.; Berg, K.C.; Eilertsen, I.A.; Eide, P.W.; Johannessen, B.; Guren, M.G.; Nesbakken, A.; Bruun, J.; Lothe, R.A.; et al. Molecular correlates of sensitivity to PARP inhibition beyond homologous recombination deficiency in pre-clinical models of colorectal cancer point to wild-type TP53 activity. EBioMedicine 2020, 59, 102923. [Google Scholar] [CrossRef] [PubMed]
- Arena, S.; Corti, G.; Durinikova, E.; Montone, M.; Reilly, N.M.; Russo, M.; Lorenzato, A.; Arcella, P.; Lazzari, L.; Rospo, G.; et al. A Subset of Colorectal Cancers with Cross-Sensitivity to Olaparib and Oxaliplatin. Clin. Cancer Res. 2020, 26, 1372–1384. [Google Scholar] [CrossRef] [PubMed]
- Kalimutho, M.; Bain, A.L.; Mukherjee, B.; Nag, P.; Nanayakkara, D.M.; Harten, S.K.; Harris, J.L.; Subramanian, G.N.; Sinha, D.; Shirasawa, S.; et al. Enhanced dependency of KRAS-mutant colorectal cancer cells on RAD51-dependent homologous recombination repair identified from genetic interactions inSaccharomyces cerevisiae. Mol. Oncol. 2017, 11, 470–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Li, X.; Li, G.; Meng, Y.; Jin, Y.; Shang, S.; Li, Y. Alpinumisoflavone causes DNA damage in Colorectal Cancer Cells via blocking DNA repair mediated by RAD51. Life Sci. 2019, 216, 259–270. [Google Scholar] [CrossRef]
- Mehta, P.; Bothra, S.J. PARP inhibitors in hereditary breast and ovarian cancer and other cancers: A review. Adv. Genet. 2021, 108, 35–80. [Google Scholar] [CrossRef]
- Messina, C.; Cattrini, C.; Soldato, D.; Vallome, G.; Caffo, O.; Castro, E.; Olmos, D.; Boccardo, F.; Zanardi, E. BRCA Mutations in Prostate Cancer: Prognostic and Predictive Implications. J. Oncol. 2020, 2020, 4986365. [Google Scholar] [CrossRef]
- Czito, B.G.; Deming, D.A.; Jameson, G.S.; Mulcahy, M.F.; Vaghefi, H.; Dudley, M.W.; Holen, K.D.; DeLuca, A.; Mittapalli, R.K.; Munasinghe, W.; et al. Safety and tolerability of veliparib combined with capecitabine plus radiotherapy in patients with locally advanced rectal cancer: A phase 1b study. Lancet Gastroenterol. Hepatol. 2017, 2, 418–426. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Slack, R.S.; Jiang, W.; He, A.R.; Hwang, J.J.; Hankin, A.; Dorsch-Vogel, K.; Kukadiya, D.; Weiner, L.M.; Marshall, J.L.; et al. A phase 2 study of the PARP inhibitor veliparib plus temozolomide in patients with heavily pretreated metastatic colorectal cancer. Cancer 2018, 124, 2337–2346. [Google Scholar] [CrossRef]
- Paviolo, N.S.; De La Vega, M.B.; Pansa, M.F.; García, I.A.; Calzetta, N.L.; Soria, G.; Gottifredi, V. Persistent double strand break accumulation does not precede cell death in an Olaparib-sensitive BRCA-deficient colorectal cancer cell model. Genet. Mol. Biol. 2020, 43, e20190070. [Google Scholar] [CrossRef] [Green Version]
- Gorbunova, V.; Beck, J.T.; Hofheinz, R.-D.; Garcia-Alfonso, P.; Nechaeva, M.; Gracian, A.C.; Mangel, L.; Fernandez, E.E.; Deming, D.A.; Ramanathan, R.K.; et al. A phase 2 randomised study of veliparib plus FOLFIRI ± bevacizumab versus placebo plus FOLFIRI ± bevacizumab in metastatic colorectal cancer. Br. J. Cancer 2018, 120, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Berlin, J.; Ramanathan, R.K.; Strickler, J.; Subramaniam, D.S.; Marshall, J.; Kang, Y.-K.; Hetman, R.; Dudley, M.W.; Zeng, J.; Nickner, C.; et al. A phase 1 dose-escalation study of veliparib with bimonthly FOLFIRI in patients with advanced solid tumours. Br. J. Cancer 2018, 118, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.M.G.; Kuznicki, A.M.; Andrade, P.; Dolinski, B.M.; Elbi, C.; O’Hagan, R.C.; Toniatti, C. Treatment with the PARP inhibitor, niraparib, sensitizes colorectal cancer cell lines to irinotecan regardless of MSI/MSS status. Cancer Cell Int. 2015, 15, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reisländer, T.; Lombardi, E.P.; Groelly, F.J.; Miar, A.; Porru, M.; Di Vito, S.; Wright, B.; Lockstone, H.; Biroccio, A.; Harris, A.; et al. BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat. Commun. 2019, 10, 3143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiong, K.-L.; Chang, K.-C.; Yeh, K.-T.; Liu, T.-Y.; Wu, J.-H.; Hsieh, P.-H.; Lin, S.-H.; Lai, W.-Y.; Hsu, Y.-C.; Chen, J.-Y.; et al. CSNK1E/CTNNB1 are synthetic lethal to TP53 in colorectal cancer and are markers for prognosis. Neoplasia 2014, 16, 441–450. [Google Scholar] [CrossRef] [Green Version]
- Makondi, P.T.; Lee, C.-H.; Huang, C.-Y.; Chu, C.-M.; Chang, Y.-J.; Wei, P.-L. Prediction of novel target genes and pathways involved in bevacizumab-resistant colorectal cancer. PLoS ONE 2018, 13, e0189582. [Google Scholar] [CrossRef]
- Schrempf, A.; Slyskova, J.; Loizou, J.I. Targeting the DNA Repair Enzyme Polymerase θ in Cancer Therapy. Trends Cancer 2020, 7, 98–111. [Google Scholar] [CrossRef]
- Soyano, A.E.; Baldeo, C.; Kasi, P.M. BRCA Mutation and Its Association with Colorectal Cancer. Clin. Color. Cancer 2018, 17, e647–e650. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Wang, Y.; Du, L.; Xu, C.; Sun, Y.; Yang, B.; Sun, Z.; Fu, Y.; Cai, L.; Fan, S.; et al. shRNA-mediated XRCC2 gene knockdown efficiently sensitizes colon tumor Cells to X-ray irradiation in vitro and in vivo. Int. J. Mol. Sci. 2014, 15, 2157–2171. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Song, X.; Chen, Z.; Qin, C. miR-7 inhibits colorectal cancer cell proliferation and induces apoptosis by targeting XRCC2. OncoTargets Ther. 2014, 7, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Andreassen, P.R.; Hanenberg, H. XRCC2 (X-ray repair cross complementing 2). Atlas Genet. Cytogenet. Oncol. Haematol. 2019, 23, 1–7. [Google Scholar] [CrossRef]
- Park, J.-Y.; Virts, E.L.; Jankowska, A.; Wiek, C.; Othman, M.; Chakraborty, S.C.; Vance, G.H.; Alkuraya, F.S.; Hanenberg, H.; Andreassen, P.R. Complementation of hypersensitivity to DNA interstrand crosslinking agents demonstrates thatXRCC2is a Fanconi anaemia gene. J. Med. Genet. 2016, 53, 672–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, K.; Song, X.; Chen, Z.; Qin, C.; He, Y.; Zhan, W. XRCC2 Promotes Colorectal Cancer Cell Growth, Regulates Cell Cycle Progression, and Apoptosis. Medicine 2014, 93, e294. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Hou, W.; Liu, X.; Chai, J.; Guo, H.; Yu, J. Targeting REV7 effectively reverses 5-FU and oxaliplatin resistance in colorectal cancer. Cancer Cell Int. 2020, 20, 580. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene(s) and No. | Study Group | Study Method | N. of FANC Gene Mutation Carriers in CRC Patients | Reference |
|---|---|---|---|---|
| FANCD1/BRCA2: 2 | 2 patients from 1 family, no mutations in known CRC genes | Point mutation screening of the BRCA1 and BRCA2 genes | 2/2 | [73] |
| FANCD1/BRCA2: 2 | 48 FCCTX probands with strong familial CRC aggregation; no mutations in known CRC genes | Mutation screening of BRCA2 | 2/48 | [85] |
| FANCD1/BRCA2: 9 FANCS/BRCA1: 6 FANCJ/BRIP1: 2 | 1260 CRC patients with suspected Lynch syndrome | 25-gene NGS panel testing | 17/1260 | [86] |
| FANCS/BRCA1: 4 FANCD1/BRCA2: 1 FANCN/PALB2: 2 | 450 EOCRC patients | 25-gene NGS panel testing | 7/450 | [30] |
| FANCS/BRCA1: 1 | 430 EOCRC patients < 50 years | 154-gene NGS panel testing | 1/430 | [87] |
| FANCI:1, FANCL: 1 FANCO/RAD51C: 1 FANCQ/ERCC4: 1 FANCS/BRCA1: 1 FANCU/XRCC2: 1 | 330 mCRC patients age ≤ 55 years, 110 mCRC patients age > 55 years | 98-gene NGS panel testing | 6/440 | [44] |
| FANCD1/BRCA2: 179 FANCS/BRCA1: 72 | 6396 unselected CRC samples | 592-gene NGS panel testing | 251/6396 | [88] |
| FANCD1/BRCA2: 8 FANCJ/BRIP1: 3 FANCS/BRCA1: 3 FANCN/PALB2: 2 | 1058 unselected CRC samples | 25-gene NGS panel testing | 16/1058 | [27] |
| FANCN/PALB2: 3 | 680 unselected CRC patients | 40-gene NGS panel testing | 3/680 | [72] |
| FANCD1/BRCA2: 1 FANCS/BRCA1: 1 | 618 unselected CRC patients | 73-gene NGS panel testing | 2/618 | [29] |
| FANCJ/BRIP1: 3 FANCD1/BRCA2: 2 FANCS/BRCA1: 2 FANCU/XRCC2: 1 | 189 unselected CRC patients | 25-gene NGS panel testing | 8/189 | [83] |
| FANCD1/BRCA2: 1 FANCN/PALB2: 1 | 88 EOCRC patients ≤ 50, MMR-proficient | WES | 2/88 | [84] |
| FANCD1/BRCA2: 4 FANCJ/BRIP1: 1 FANCO/RAD51C: 1 | 133 EOCRC patients < 55 years | WES | 6/133 | [89] |
| FANCD1/BRCA2: 2 FANCC: 1, FANCE: 1 FANCJ/BRIP1: 1 | 74 CRC patients from 40 unrelated families with strong CRC aggregation; no mutations in known CRC genes | WES | 5/74 | [28] |
| FANCM: 4 | 94 CRC patients (47 CRC-affected cousin pairs) | WES | 4/94 | [96] |
| FANCA: 1 FANCD1/BRCA2: 1 FANCD2: 1, FANCM: 2 | 141 unselected CRC patients | WES | 5/141 | [31] |
| FA Component | FA Component Status | Drug Treatment(s) or Synthetic Lethality Partner(s) | Study Materials | Setting | Mechanism of Action/Observed Results | Ref. |
|---|---|---|---|---|---|---|
| FANCR/RAD51 | RAD51 inhibition (B02 inhibitor) | Prexasertib (CHK1/2 inhibitors) | CRC stem cells | In vitro | Triggering mitotic catastrophe | [131] |
| RAD51 inhibition (B02 inhibitor) | Mirin (MRE11 inhibitor) | PARP1-upregulated CRC stem cells | In vitro and in vivo | Triggering mitotic catastrophe | [129] | |
| Decreased RAD51 protein | Talazoparib (PARP inhibitor) | TP53 wild-type cell lines | In vitro | Increased sensitivity to the PARP inhibitor | [132] | |
| Increased RAD51 foci formation | Olaparib (PARP inhibitor) | Patient-derived CRC models | In vitro | Resistance to the PARP inhibitor | [133] | |
| RAD51 inhibition (RI-1 inhibitor) | AZD6244 (MEK1/2 inhibitor) | KRAS-mutant cells | In vitro | Induction of DNA damage and apoptosis | [134] | |
| RAD51 knockdown (specific siRNA) | Alpinumisoflavone (natural flavonoid) | CRC cell lines | In vitro | Increased anti-cancer activity of alpinumisoflavone | [135] | |
| FANCD1/S (BRCA1/2) | BRCA1/2 depletion (specific shRNA) | Olaparib (PARP inhibitor) | BRCA-deficient cell lines | In vitro | Genomic instability and cell death | [140] |
| BRCA1/2 depletion (specific shRNA) | Olaparib and talazoparib (PARP inhibitors) | BRCA-deficient cell lines | In vitro and in vivo | Elicit innate immune response | [144] | |
| ND | Niraparib (PARP inhibitor) and irinotecan | MSI or MSS CRC cells | In vitro or in vivo | Enhancement of the anti-tumor effects of both agents | [143] | |
| BRCA1 gene expression | POLB, CSNK1E, MSH2 | GEO datasets-mCRC patients and CRC cells | Translational and in vitro | Synthetic lethality | [145] | |
| BRCA2 gene expression | MSH2 | GEO datasets-mCRC patients and CRC cells | Translational and in vitro | Synthetic lethality | [145] | |
| High BRCA1 gene expression | Bevacizumab (VEGF Inhibitor) | GEO datasets-mCRC patients | Translational | More favourable PFS | [146] | |
| Mutated BRCA1 | Oxaliplatin plus radiation before surgery | One LARC patient | Case report | Increased sensitivity to platinum-based chemotherapy | [148] | |
| FANCU/XRCC2 | XRCC2 depletion (specific shRNA) | X-radiation | T84 colon tumor cell line | In vitro and In vivo | Increased sensitivity to X-radiation | [149] |
| XRCC2 targeting by miR-7 | - | CRC cell lines | In vitro | Apoptosis and inhibition of proliferation | [151] | |
| Biallelic mutated XRCC2 | Olaparib (PARP inhibitor) | Fibroblast cells | In vitro | Increased sensitivity to olaparib | [152] | |
| Increased expression of XRCC2 | Olaparib (PARP inhibitor) | CRC cell lines | In vitro | Synthetic lethality | [153] | |
| FANCV/REV7 | REV7 depletion (CRISPR/Cas9) | 5-fluorouracil and oxaliplatin | CRC cells | In vitro and In vivo | Impair of translesion DNA synthesis pathway | [154] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parsa, F.G.; Nobili, S.; Karimpour, M.; Aghdaei, H.A.; Nazemalhosseini-Mojarad, E.; Mini, E. Fanconi Anemia Pathway in Colorectal Cancer: A Novel Opportunity for Diagnosis, Prognosis and Therapy. J. Pers. Med. 2022, 12, 396. https://doi.org/10.3390/jpm12030396
Parsa FG, Nobili S, Karimpour M, Aghdaei HA, Nazemalhosseini-Mojarad E, Mini E. Fanconi Anemia Pathway in Colorectal Cancer: A Novel Opportunity for Diagnosis, Prognosis and Therapy. Journal of Personalized Medicine. 2022; 12(3):396. https://doi.org/10.3390/jpm12030396
Chicago/Turabian StyleParsa, Fatemeh Ghorbani, Stefania Nobili, Mina Karimpour, Hamid Asadzadeh Aghdaei, Ehsan Nazemalhosseini-Mojarad, and Enrico Mini. 2022. "Fanconi Anemia Pathway in Colorectal Cancer: A Novel Opportunity for Diagnosis, Prognosis and Therapy" Journal of Personalized Medicine 12, no. 3: 396. https://doi.org/10.3390/jpm12030396