
Computational Models for Clinical Applications in Personalized Medicine—Guidelines and Recommendations for Data Integration and Model Validation

 ,
,
Abstract
:1. Introduction
2. Modelling Approaches for Clinical Applications in Personalized Medicine
2.1. Mechanistic Models
2.1.1. Molecular Interaction Maps
2.1.2. Constraint-Based Models
2.1.3. Boolean Models
2.1.4. Quantitative Models
2.1.5. Pharmacokinetic Models
2.1.6. Software Resources and Tools
2.2. Machine Learning and Deep Learning
3. Models in Clinical Research for Discovery, Diagnosis, and Therapy
3.1. Discovery
3.2. Diagnosis
3.3. Therapy
4. Challenges and Recommendations
4.1. Challenges
4.1.1. Data Availability and Data Harmonization
4.1.2. Model Development and Model Validation
4.1.3. Model Standardization, Model Re-use, and Reporting of Results
4.1.4. Legal and Ethical Issues
4.2. Recommendations
4.2.1. Study Design
4.2.2. Data Acquisition and Operation
4.2.3. Model Development and Model Validation
4.2.4. Translations and Applications
5. Conclusions
- Careful planning of study design is of utmost importance at the project start;
- Common standards for data sampling, data acquisition, and data operation should be fulfilled;
- Data harmonization is crucial to ensure data compatibility and comparability;
- Data should be divided in data sets for training and validation;
- Model documentation should be written according to best practice guidelines;
- It is important to openly communicate model assumptions and biases in the computational results;
- New patient data should be continuously used for benchmarking of the computational results.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wolkenhauer, O.; Auffray, C.; Brass, O.; Clairambault, J.; Deutsch, A.; Drasdo, D.; Gervasio, F.; Preziosi, L.; Maini, P.; Marciniak-Czochra, A.; et al. Enabling multiscale modeling in systems medicine. Genome Med. 2014, 6, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apweiler, R.; Beissbarth, T.; Berthold, M.R.; Bluthgen, N.; Burmeister, Y.; Dammann, O.; Deutsch, A.; Feuerhake, F.; Franke, A.; Hasenauer, J.; et al. Whither systems medicine? Exp. Mol. Med. 2018, 50, e453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pison, C.; The CASyM consortium. The CASyM roadmap-Implementation of Systems Medicine across Europe. 2014. Available online: www.casym.eu (accessed on 18 November 2021).
- Morrison, T.M.; Pathmanathan, P.; Adwan, M.; Margerrison, E. Advancing Regulatory Science with Computational Modeling for Medical Devices at the FDA’s Office of Science and Engineering Laboratories. Front. Med. 2018, 5, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musuamba, F.T.; Skottheim Rusten, I.; Lesage, R.; Russo, G.; Bursi, R.; Emili, L.; Wangorsch, G.; Manolis, E.; Karlsson, K.E.; Kulesza, A.; et al. Scientific and Regulatory Evaluation of Mechanistic In Silico Drug and Disease Models in Drug Development: Building Model Credibility. CPT Pharmacomet. Syst. Pharmacol. 2021, 10, 804–825. [Google Scholar] [CrossRef]
- EMA. Guideline on the Reporting of Physiologically Based Pharmacokinetic (PBPK) Modelling and Simulation; European Medicines Agency: Amsterdam, The Netherlands, 2018.
- Morrison, T.M. Reporting of Computational Modeling Studies in Medical Device Submissions Guidance for Industry and Food and Drug Administration Staff; O.o.S.a.E. Laboratories, Ed.; U.S. Department of Health and Human Services Food and Drug Administration: Silver Spring, MD, USA, 2016.
- Barredo Arrieta, A.; Díaz-Rodríguez, N.; Del Ser, J.; Bennetot, A.; Tabik, S.; Barbado, A.; Garcia, S.; Gil-Lopez, S.; Molina, D.; Benjamins, R.; et al. Explainable Artificial Intelligence (XAI): Concepts, taxonomies, opportunities and challenges toward responsible AI. Inf. Fusion 2020, 58, 82–115. [Google Scholar] [CrossRef] [Green Version]
- Dimiduk, D.M.; Holm, E.A.; Niezgoda, S.R. Perspectives on the Impact of Machine Learning, Deep Learning, and Artificial Intelligence on Materials, Processes, and Structures Engineering. Integr. Mater. Manuf. Innov. 2018, 7, 157–172. [Google Scholar] [CrossRef] [Green Version]
- Wolkenhauer, O. Why model? Front. Physiol. 2014, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Wolkenhauer, O.; Auffray, C.; Jaster, R.; Steinhoff, G.; Dammann, O. The road from systems biology to systems medicine. Pediatr. Res. 2013, 73, 502–507. [Google Scholar] [CrossRef] [Green Version]
- Wolkenhauer, O.; Green, S. The search for organizing principles as a cure against reductionism in systems medicine. FEBS J. 2013, 280, 5938–5948. [Google Scholar] [CrossRef] [Green Version]
- Kitano, H.; Funahashi, A.; Matsuoka, Y.; Oda, K. Using process diagrams for the graphical representation of biological networks. Nat. Biotechnol. 2005, 23, 961–966. [Google Scholar] [CrossRef]
- Fujita, K.A.; Ostaszewski, M.; Matsuoka, Y.; Ghosh, S.; Glaab, E.; Trefois, C.; Crespo, I.; Perumal, T.M.; Jurkowski, W.; Antony, P.M.; et al. Integrating pathways of Parkinson’s disease in a molecular interaction map. Mol. Neurobiol. 2014, 49, 88–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuperstein, I.; Bonnet, E.; Nguyen, H.A.; Cohen, D.; Viara, E.; Grieco, L.; Fourquet, S.; Calzone, L.; Russo, C.; Kondratova, M.; et al. Atlas of Cancer Signalling Network: A Systems Biology Resource for Integrative Analysis of Cancer Data with Google Maps. Oncogenesis 2015, 4, e160. [Google Scholar] [CrossRef] [Green Version]
- Thiele, I.; Palsson, B.O. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010, 5, 93–121. [Google Scholar] [CrossRef] [Green Version]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Nookaew, I.; Jacobson, P.; Walley, A.J.; Froguel, P.; Carlsson, L.M.; Uhlen, M.; et al. Integration of clinical data with a genome-scale metabolic model of the human adipocyte. Mol. Syst. Biol. 2013, 9, 649. [Google Scholar] [CrossRef] [PubMed]
- Stempler, S.; Yizhak, K.; Ruppin, E. Integrating transcriptomics with metabolic modeling predicts biomarkers and drug targets for Alzheimer’s disease. PLoS ONE 2014, 9, e105383. [Google Scholar]
- Wang, S.R.; Saadatpour, A.; Albert, R. Boolean modeling in systems biology: An overview of methodology and applications. Phys. Biol. 2012, 9, 055001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eduati, F.; Jaaks, P.; Wappler, J.; Cramer, T.; Merten, C.A.; Garnett, M.J.; Saez-Rodriguez, J. Patient-specific logic models of signaling pathways from screenings on cancer biopsies to prioritize personalized combination therapies. Mol. Syst. Biol. 2020, 16, e8664. [Google Scholar] [CrossRef]
- Udyavar, A.R.; Wooten, D.J.; Hoeksema, M.; Bansal, M.; Califano, A.; Estrada, L.; Schnell, S.; Irish, J.M.; Massion, P.P.; Quaranta, V. Correction: Novel Hybrid. Phenotype Revealed in Small Cell Lung Cancer by a Transcription Factor Network Model. That Can Explain Tumor Heterogeneity. Cancer Res. 2019, 79, 1014. [Google Scholar] [CrossRef] [Green Version]
- Malik-Sheriff, R.S.; Glont, M.; Nguyen, T.V.N.; Tiwari, K.; Roberts, M.G.; Xavier, A.; Vu, M.T.; Men, J.; Maire, M.; Kananathan, S.; et al. BioModels-15 years of sharing computational models in life science. Nucleic Acids Res. 2020, 48, D407–D415. [Google Scholar] [CrossRef] [Green Version]
- Kolch, W.; Fey, D. Personalized Computational Models as Biomarkers. J. Pers. Med. 2017, 7, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hastings, J.F.; O’Donnell, Y.E.I.; Fey, D.; Croucher, D.R. Applications of personalised signalling network models in precision oncology. Pharmacol. Ther. 2020, 212, 107555. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Urizar, J.; Granados-Soto, V.; Flores-Murrieta, F.J.; Castañeda-Hernández, G. Pharmacokinetic-Pharmacodynamic Modeling: Why? Arch. Med. Res. 2000, 31, 539–545. [Google Scholar] [CrossRef]
- Edginton, N.A.; Willmann, S. Physiology-based simulations of a pathological condition: Prediction of pharmacokinetics in patients with liver cirrhosis. Clin. Pharmacokinet. 2008, 47, 743–752. [Google Scholar] [CrossRef]
- Maharaj, R.A.; Edginton, A.N. Physiologically based pharmacokinetic modeling and simulation in pediatric drug development. CPT Pharmacomet. Syst. Pharm. 2014, 3, e150. [Google Scholar] [CrossRef] [Green Version]
- Jones, H.M.; Chen, Y.; Gibson, C.; Heimbach, T.; Parrott, N.; Peters, S.A.; Snoeys, J.; Upreti, V.V.; Zheng, M.; Hall, S.D. Physiologically based pharmacokinetic modeling in drug discovery and development: A pharmaceutical industry perspective. Clin. Pharmacol. Ther. 2015, 97, 247–262. [Google Scholar] [CrossRef]
- Swat, M.J.; Moodie, S.; Wimalaratne, S.M.; Kristensen, N.R.; Lavielle, M.; Mari, A.; Magni, P.; Smith, M.K.; Bizzotto, R.; Pasotti, L.; et al. Pharmacometrics Markup Language (PharmML): Opening New Perspectives for Model. Exchange in Drug Development. CPT Pharmacomet. Syst. Pharm. 2015, 4, 316–319. [Google Scholar] [CrossRef]
- Kuepfer, L.; Niederalt, C.; Wendl, T.; Schlender, J.F.; Willmann, S.; Lippert, J.; Block, M.; Eissing, T.; Teutonico, D. Applied Concepts in PBPK Modeling: How to Build. a PBPK/PD Model. CPT Pharmacomet. Syst. Pharm. 2016, 5, 516–531. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, T.; Scotcher, D.; Darwich, A.S.; Galetin, A.; Rostami-Hodjegan, A. Towards Further Verification of Physiologically-Based Kidney Models: Predictability of the Effects of Urine-Flow and Urine-pH on Renal Clearance. J. Pharmacol. Exp. Ther. 2019, 368, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Schlender, J.F.; Meyer, M.; Thelen, K.; Krauss, M.; Willmann, S.; Eissing, T.; Jaehde, U. Development of a Whole-Body Physiologically Based Pharmacokinetic Approach to Assess. the Pharmacokinetics of Drugs in Elderly Individuals. Clin. Pharm. 2016, 55, 1573–1589. [Google Scholar]
- Maharaj, R.A.; Edginton, A.N.; Fotaki, N. Assessment of Age-Related Changes in Pediatric Gastrointestinal Solubility. Pharm. Res. 2016, 33, 52–71. [Google Scholar] [CrossRef] [PubMed]
- Turei, D.; Korcsmaros, T.; Saez-Rodriguez, J. OmniPath: Guidelines and gateway for literature-curated signaling pathway resources. Nat. Methods 2016, 13, 966–967. [Google Scholar] [CrossRef] [PubMed]
- Politano, G.; di Carlo, S.; Benso, A. ’One DB to rule them all’-the RING: A Regulatory Interaction Graph. combining TFs, genes/proteins, SNPs, diseases and drugs. Database 2019, 2019, baz108. [Google Scholar] [CrossRef] [Green Version]
- Klamt, S.; Saez-Rodriguez, J.; Gilles, E.D. Structural and functional analysis of cellular networks with CellNetAnalyzer. BMC Syst. Biol. 2007, 1, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helikar, T.; Kowal, B.; McClenathan, S.; Bruckner, M.; Rowley, T.; Madrahimov, A.; Wicks, B.; Shrestha, M.; Limbu, K.; Rogers, J.A. The Cell Collective: Toward an Open and Collaborative Approach to Systems Biology. BMC Syst. Biol. 2012, 6, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naldi, A.; Berenguier, D.; Faure, A.; Lopez, F.; Thieffry, D.; Chaouiya, C. Logical modelling of regulatory networks with GINsim 2.3. Biosystems 2009, 97, 134–139. [Google Scholar] [CrossRef]
- Licata, L.; Lo Surdo, P.; Iannuccelli, M.; Palma, A.; Micarelli, E.; Perfetto, L.; Peluso, D.; Calderone, A.; Castagnoli, L.; Cesareni, G. SIGNOR 2.0, the SIGnaling Network Open Resource 2.0: 2019 update. Nucleic Acids Res. 2020, 48, D504–D510. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Csabai, L.; Fazekas, D.; Kadlecsik, T.; Szalay-Beko, M.; Bohar, B.; Madgwick, M.; Modos, D.; Olbei, M.; Gul, L.; Sudhakar, P.; et al. SignaLink3: A multi-layered resource to uncover tissue-specific signaling networks. Nucleic Acids Res. 2022, 50, D701–D709. [Google Scholar] [CrossRef]
- Breuer, K.; Foroushani, A.K.; Laird, M.R.; Chen, C.; Sribnaia, A.; Lo, R.; Winsor, G.L.; Hancock, R.E.; Brinkman, F.S.; Lynn, D.J. InnateDB: Systems biology of innate immunity and beyond-Recent updates and continuing curation. Nucleic Acids Res. 2013, 41, D1228–D1233. [Google Scholar] [CrossRef]
- Slenter, D.N.; Kutmon, M.; Hanspers, K.; Riutta, A.; Windsor, J.; Nunes, N.; Melius, J.; Cirillo, E.; Coort, S.L.; Digles, D.; et al. WikiPathways: A multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res. 2018, 46, D661–D667. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mussel, C.; Hopfensitz, M.; Kestler, H.A. BoolNet-An R package for generation, reconstruction and analysis of Boolean networks. Bioinformatics 2010, 26, 1378–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glont, M.; Nguyen, T.V.N.; Graesslin, M.; Halke, R.; Ali, R.; Schramm, J.; Wimalaratne, S.M.; Kothamachu, V.B.; Rodriguez, N.; Swat, M.J.; et al. BioModels: Expanding horizons to include more modelling approaches and formats. Nucleic Acids Res. 2018, 46, D1248–D1253. [Google Scholar] [CrossRef]
- De Jong, H.; Geiselmann, J.; Hernandez, C.; Page, M. Genetic Network Analyzer: Qualitative simulation of genetic regulatory networks. Bioinformatics 2003, 19, 336–344. [Google Scholar] [CrossRef] [Green Version]
- Di Cara, A.; Garg, A.; De Micheli, G.; Xenarios, I.; Mendoza, L. Dynamic simulation of regulatory networks using SQUAD. BMC Bioinform. 2007, 8, 462. [Google Scholar] [CrossRef] [Green Version]
- Stoll, G.; Caron, B.; Viara, E.; Dugourd, A.; Zinovyev, A.; Naldi, A.; Kroemer, G.; Barillot, E.; Calzone, L. MaBoSS 2.0: An environment for stochastic Boolean modeling. Bioinformatics 2017, 33, 2226–2228. [Google Scholar] [CrossRef] [Green Version]
- Terfve, C.; Cokelaer, T.; Henriques, D.; MacNamara, A.; Goncalves, E.; Morris, M.K.; Van Iersel, M.; Lauffenburger, D.A.; Saez-Rodriguez, J. CellNOptR: A flexible toolkit to train protein signaling networks to data using multiple logic formalisms. BMC Syst. Biol. 2012, 6, 133. [Google Scholar] [CrossRef] [Green Version]
- King, Z.A.; Lu, J.; Drager, A.; Miller, P.; Federowicz, S.; Lerman, J.A.; Ebrahim, A.; Palsson, B.O.; Lewis, N.E. BiGG Models: A platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res. 2016, 44, D515–D522. [Google Scholar] [CrossRef]
- Pornputtapong, N.; Nookaew, I.; Nielsen, J. Human metabolic atlas: An online resource for human metabolism. Database 2015, 2015, bav068. [Google Scholar] [CrossRef]
- Noronha, A.; Modamio, J.; Jarosz, Y.; Guerard, E.; Sompairac, N.; Preciat, G.; Danielsdottir, A.D.; Krecke, M.; Merten, D.; Haraldsdottir, H.S.; et al. The Virtual Metabolic Human Database: Integrating Human and Gut Microbiome Metabolism with Nutrition and Disease. Nucleic Acids Res. 2019, 47, D614–D624. [Google Scholar] [CrossRef] [PubMed]
- Schellenberger, J.; Que, R.; Fleming, R.M.; Thiele, I.; Orth, J.D.; Feist, A.M.; Zielinski, D.C.; Bordbar, A.; Lewis, N.E.; Rahmanian, S.; et al. Quantitative prediction of cellular metabolism with constraint-based models: The COBRA Toolbox v2.0. Nat. Protoc. 2011, 6, 1290–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelius-Dietrich, G.; Desouki, A.A.; Fritzemeier, C.J.; Lercher, M.J. Sybil-Efficient constraint-based modelling in R. BMC Syst. Biol. 2013, 7, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebrahim, A.; Lerman, J.A.; Palsson, B.O.; Hyduke, D.R. COBRApy: Constraints-Based Reconstruction and Analysis for Python. BMC Syst. Biol. 2013, 7, 74. [Google Scholar] [CrossRef] [Green Version]
- Seaver, S.M.D.; Liu, F.; Zhang, Q.; Jeffryes, J.; Faria, J.P.; Edirisinghe, J.N.; Mundy, M.; Chia, N.; Noor, E.; Beber, M.E.; et al. The ModelSEED Biochemistry Database for the Integration of Metabolic Annotations and the Reconstruction, Comparison and Analysis of Metabolic Models for Plants, Fungi and Microbes. Nucleic Acids Res. 2021, 49, D575–D588. [Google Scholar] [CrossRef]
- Chelliah, V.; Laibe, C.; le Novere, N. BioModels Database: A Repository of Mathematical Models of Biological Processes. Methods Mol. Biol. 2013, 1021, 189–199. [Google Scholar]
- Peters, M.; Eicher, J.J.; Van Niekerk, D.D.; Waltemath, D.; Snoep, J.L. The JWS online simulation database. Bioinformatics 2017, 33, 1589–1590. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Lloyd, C.M.; Nickerson, D.P.; Cooling, M.T.; Miller, A.K.; Garny, A.; Terkildsen, J.R.; Lawson, J.; Britten, R.D.; Hunter, P.J.; et al. The Physiome Model Repository 2. Bioinformatics 2011, 27, 743–744. [Google Scholar] [CrossRef] [Green Version]
- Hoops, S.; Sahle, S.; Gauges, R.; Lee, C.; Pahle, J.; Simus, N.; Singhal, M.; Xu, L.; Mendes, P.; Kummer, U. COPASI-A Complex Pathway Simulator. Bioinformatics 2006, 22, 3067–3074. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, Y.; Funahashi, A.; Ghosh, S.; Kitano, H. Modeling and simulation using CellDesigner. Methods Mol. Biol. 2014, 1164, 121–145. [Google Scholar]
- Hucka, M.; Nickerson, D.P.; Bader, G.D.; Bergmann, F.T.; Cooper, J.; Demir, E.; Garny, A.; Golebiewski, M.; Myers, C.J.; Schreiber, F.; et al. Promoting Coordinated Development of Community-Based Information Standards for Modeling in Biology: The COMBINE Initiative. Front. Bioeng. Biotechnol. 2015, 3, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hucka, M.; Bergmann, F.T.; Chaouiya, C.; Drager, A.; Hoops, S.; Keating, S.M.; Konig, M.; Novere, N.L.; Myers, C.J.; Olivier, B.G.; et al. The Systems Biology Markup Language (SBML): Language Specification for Level 3 Version 2 Core Release 2. Mol. Syst. Biol. 2020, 16, e9110. [Google Scholar]
- Clerx, M.; Cooling, M.T.; Cooper, J.; Garny, A.; Moyle, K.; Nickerson, D.P.; Nielsen, P.M.F.; Sorby, H. CellML 2.0. J. Integr. Bioinform. 2020, 17, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Le Novere, N.; Hucka, M.; Mi, H.; Moodie, S.; Schreiber, F.; Sorokin, A.; Demir, E.; Wegner, K.; Aladjem, M.I.; Wimalaratne, S.M.; et al. The Systems Biology Graphical Notation. Nat. Biotechnol. 2009, 27, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Mazein, A.; Ostaszewski, M.; Kuperstein, I.; Watterson, S.; Le Novere, N.; Lefaudeux, D.; De Meulder, B.; Pellet, J.; Balaur, I.; Saqi, M.; et al. Systems medicine disease maps: Community-driven comprehensive representation of disease mechanisms. NPJ Syst. Biol. Appl. 2018, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Chaouiya, C.; Berenguier, D.; Keating, S.M.; Naldi, A.; Van Iersel, M.P.; Rodriguez, N.; Drager, A.; Buchel, F.; Cokelaer, T.; Kowal, B.; et al. SBML qualitative models: A model representation format and infrastructure to foster interactions between qualitative modelling formalisms and tools. BMC Syst. Biol. 2013, 7, 135. [Google Scholar] [CrossRef] [Green Version]
- Mourby, M.; Cathaoir, K.Ó.; Collin, C.B. Transparency of machine-learning in healthcare: The GDPR & European health law. Comput. Law Secur. Rev. 2021, 43, 105611. [Google Scholar]
- Thorsen-Meyer, H.-C.; Nielsen, A.B.; Nielsen, A.P.; Kaas-Hansen, B.S.; Toft, P.; Schierbeck, J.; Strøm, T.; Chmura, P.J.; Heimann, M.; Dybdahl, L.; et al. Dynamic and explainable machine learning prediction of mortality in patients in the intensive care unit: A retrospective study of high-frequency data in electronic patient records. Lancet Digit. Health 2020, 2, e179–e191. [Google Scholar] [CrossRef]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Kim, J.; Kim, J.; Jang, G.J.; Lee, M. Fast learning method for convolutional neural networks using extreme learning machine and its application to lane detection. Neural Netw. 2017, 87, 109–121. [Google Scholar] [CrossRef]
- Greener, J.G.; Kandathil, S.M.; Moffat, L.; Jones, D.T. A guide to machine learning for biologists. Nat. Rev. Mol. Cell Biol. 2021, 23, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Weissler, E.H.; Naumann, T.; Andersson, T.; Ranganath, R.; Elemento, O.; Luo, Y.; Freitag, D.F.; Benoit, J.; Hughes, M.C.; Khan, F.; et al. The role of machine learning in clinical research: Transforming the future of evidence generation. Trials 2021, 22, 537. [Google Scholar] [CrossRef]
- Beaulieu-Jones, B.K.; Yuan, W.; Brat, G.A.; Beam, A.L.; Weber, G.; Ruffin, M.; Kohane, I.S. Machine learning for patient risk stratification: Standing on, or looking over, the shoulders of clinicians? NPJ Digit. Med. 2021, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Baldi, P.; Brunak, S. Bioinformatics: The Machine Learning Approach; MIT Press: Cambridge, MA, USA, 2001. [Google Scholar]
- Liu, X.; Faes, L.; Kale, A.U.; Wagner, S.K.; Fu, D.J.; Bruynseels, A.; Mahendiran, T.; Moraes, G.; Shamdas, M.; Kern, C.; et al. A comparison of deep learning performance against health-care professionals in detecting diseases from medical imaging: A systematic review and meta-analysis. Lancet Digit. Health 2019, 1, e271–e297. [Google Scholar] [CrossRef]
- Stupple, A.; Singerman, D.; Celi, L.A. The reproducibility crisis in the age of digital medicine. NPJ Digit. Med. 2019, 2, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Cruz Rivera, S.; Moher, D.; Calvert, M.J.; Denniston, A.K.; The SPIRIT-AI and CONSORT-AI Working Group. Reporting guidelines for clinical trial reports for interventions involving artificial intelligence: The CONSORT-AI extension. Nat. Med. 2020, 26, 1364–1374. [Google Scholar] [CrossRef]
- Cruz Rivera, S.; Liu, X.; Chan, A.-W.; Denniston, A.K.; Calvert, M.J.; SPIRIT-AI and CONSORT-AI Working Group; SPIRIT-AI and CONSORT-AI Steering Group; SPIRIT-AI and CONSORT-AI Consensus Group. Guidelines for clinical trial protocols for interventions involving artificial intelligence: The SPIRIT-AI extension. Nat. Med. 2020, 26, 1351–1363. [Google Scholar] [CrossRef]
- Matschinske, J.; Alcaraz, N.; Benis, A.; Golebiewski, M.; Grimm, D.G.; Heumos, L.; Kacprowski, T.; Lazareva, O.; List, M.; Louadi, Z.; et al. The AIMe registry for artificial intelligence in biomedical research. Nat. Methods 2021, 18, 1128–1131. [Google Scholar] [CrossRef]
- Serhan, C.N.; Gupta, S.K.; Perretti, M.; Godson, C.; Brennan, E.; Li, Y.; Soehnlein, O.; Shimizu, T.; Werz, O.; Chiurchiu, V.; et al. The Atlas of Inflammation Resolution (AIR). Mol. Asp. Med. 2020, 74, 100894. [Google Scholar] [CrossRef]
- Wu, G.; Zhu, L.; Dent, J.E.; Nardini, C. A comprehensive molecular interaction map for rheumatoid arthritis. PLoS ONE 2010, 5, e10137. [Google Scholar] [CrossRef]
- Mazein, A.; Knowles, R.G.; Adcock, I.; Chung, K.F.; Wheelock, C.E.; Maitland-Van Der Zee, A.H.; Sterk, P.J.; Auffray, C.; AsthmaMap Project, T. AsthmaMap: An expert-driven computational representation of disease mechanisms. Clin. Exp. Allergy 2018, 48, 916–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parton, A.; McGilligan, V.; Chemaly, M.; O’Kane, M.; Watterson, S. New models of atherosclerosis and multi-drug therapeutic interventions. Bioinformatics 2019, 35, 2449–2457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varemo, L.; Scheele, C.; Broholm, C.; Mardinoglu, A.; Kampf, C.; Asplund, A.; Nookaew, I.; Uhlen, M.; Pedersen, B.K.; Nielsen, J. Proteome-and transcriptome-driven reconstruction of the human myocyte metabolic network and its use for identification of markers for diabetes. Cell Rep. 2015, 11, 921–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Uhlen, M.; Nielsen, J. Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 3083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Cerda, M.L.; Irurzun-Arana, I.; Gonzalez-Garcia, I.; Hu, C.; Zhou, H.; Vermeulen, A.; Troconiz, I.F.; Gomez-Mantilla, J.D. Towards patient stratification and treatment in the autoimmune disease lupus erythematosus using a systems pharmacology approach. Eur. J. Pharm. Sci. 2016, 94, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.M.; Marquardt, S.; Gupta, S.K.; Knoll, S.; Schmitz, U.; Spitschak, A.; Engelmann, D.; Vera, J.; Wolkenhauer, O.; Putzer, B.M. Unraveling a tumor type-specific regulatory core underlying E2F1-mediated epithelial-mesenchymal transition to predict receptor protein signatures. Nat. Commun. 2017, 8, 198. [Google Scholar] [CrossRef] [Green Version]
- Agren, R.; Mardinoglu, A.; Asplund, A.; Kampf, C.; Uhlen, M.; Nielsen, J. Identification of anticancer drugs for hepatocellular carcinoma through personalized genome-scale metabolic modeling. Mol. Syst. Biol. 2014, 10, 721. [Google Scholar] [CrossRef]
- Fey, D.; Halasz, M.; Dreidax, D.; Kennedy, S.P.; Hastings, J.F.; Rauch, N.; Munoz, A.G.; Pilkington, R.; Fischer, M.; Westermann, F.; et al. Signaling pathway models as biomarkers: Patient-specific simulations of JNK activity predict the survival of neuroblastoma patients. Sci. Signal. 2015, 8, ra130. [Google Scholar] [CrossRef] [Green Version]
- Hector, S.; Rehm, M.; Schmid, J.; Kehoe, J.; McCawley, N.; Dicker, P.; Murray, F.; McNamara, D.; Kay, E.W.; Concannon, C.G.; et al. Clinical application of a systems model of apoptosis execution for the prediction of colorectal cancer therapy responses and personalisation of therapy. Gut 2012, 61, 725–733. [Google Scholar] [CrossRef]
- Velickovski, F.; Ceccaroni, L.; Roca, J.; Burgos, F.; Galdiz, J.B.; Marina, N.; Lluch-Ariet, M. Clinical Decision Support. Systems (CDSS) for preventive management of COPD patients. J. Transl. Med. 2014, 12, S9. [Google Scholar]
- Sutton, R.T.; Pincock, D.; Baumgart, D.C.; Sadowski, D.C.; Fedorak, R.N.; Kroeker, K.I. An overview of clinical decision support systems: Benefits, risks, and strategies for success. NPJ Digit. Med. 2020, 3, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, D.M.; Friedensohn, S.; Weber, C.R.; Jordi, C.; Wagner, B.; Meng, S.M.; Ehling, R.A.; Bonati, L.; Dahinden, J.; Gainza, P.; et al. Optimization of therapeutic antibodies by predicting antigen specificity from antibody sequence via deep learning. Nat. Biomed. Eng. 2021, 5, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, C.R.; Covington, B.C.; Derewacz, D.K.; McNees, C.R.; Wikswo, J.P.; McLean, J.A.; Bachmann, B.O. Structuring Microbial Metabolic Responses to Multiplexed Stimuli via Self-Organizing Metabolomics Maps. Chem. Biol. 2015, 22, 661–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babu, R.V.; Bar, Y.; Blair, J.; Bradley, A.P.; Carneiro, G.; Chen, H.; Dam, E.B.; Diamant, I.; Dou, Q.; Feldman, M.D.; et al. Contributors. In Deep Learning for Medical Image Analysis; Zhou, S.K., Greenspan, H., Shen, D., Eds.; Academic Press: Cambridge, MA, USA, 2017. [Google Scholar]
- Chan, H.P.; Samala, R.K.; Hadjiiski, L.M.; Zhou, C. Deep Learning in Medical Image Analysis. Adv. Exp. Med. Biol. 2020, 1213, 3–21. [Google Scholar]
- Abràmoff, M.D.; Lavin, P.T.; Birch, M.; Shah, N.; Folk, J.C. Pivotal trial of an autonomous AI-based diagnostic system for detection of diabetic retinopathy in primary care offices. NPJ Digit. Med. 2018, 1, 39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Liu, X.; Shen, J.; Li, Z.; Sang, Y.; Wu, X.; Zha, Y.; Liang, W.; Wang, C.; Wang, K.; et al. Clinically Applicable AI System for Accurate Diagnosis, Quantitative Measurements, and Prognosis of COVID-19 Pneumonia Using Computed Tomography. Cell 2020, 181, 1423–1433.e11. [Google Scholar] [CrossRef] [PubMed]
- Damask, A.; Steg, P.G.; Schwartz, G.G.; Szarek, M.; Hagstrom, E.; Badimon, L.; Chapman, M.J.; Boileau, C.; Tsimikas, S.; Ginsberg, H.N.; et al. Patients with High. Genome-Wide Polygenic Risk Scores for Coronary Artery Disease May Receive Greater Clinical Benefit from Alirocumab Treatment in the ODYSSEY OUTCOMES Trial. Circulation 2020, 141, 624–636. [Google Scholar] [CrossRef]
- Duncan, L.; Shen, H.; Gelaye, B.; Meijsen, J.; Ressler, K.; Feldman, M.; Peterson, R.; Domingue, B. Analysis of polygenic risk score usage and performance in diverse human populations. Nat. Commun. 2019, 10, 3328. [Google Scholar] [CrossRef]
- Marquez-Luna, C.; Loh, P.R.; South Asian Type 2 Diabetes (SAT2D) Consortium; SIGMA Type 2 Diabetes Consortium; Price, A.L. Multiethnic polygenic risk scores improve risk prediction in diverse populations. Genet. Epidemiol. 2017, 41, 811–823. [Google Scholar] [CrossRef]
- Grinde, K.E.; Qi, Q.; Thornton, T.A.; Liu, S.; Shadyab, A.H.; Chan, K.H.K.; Reiner, A.P.; Sofer, T. Generalizing polygenic risk scores from Europeans to Hispanics/Latinos. Genet. Epidemiol. 2019, 43, 50–62. [Google Scholar] [CrossRef]
- Pare, G.; Mao, S.; Deng, W.Q. A machine-learning heuristic to improve gene score prediction of polygenic traits. Sci. Rep. 2017, 7, 12665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilhjalmsson, B.J.; Yang, J.; Finucane, H.K.; Gusev, A.; Lindstrom, S.; Ripke, S.; Genovese, G.; Loh, P.R.; Bhatia, G.; Do, R.; et al. Modeling Linkage Disequilibrium Increases Accuracy of Polygenic Risk Scores. Am. J. Hum. Genet. 2015, 97, 576–592. [Google Scholar] [CrossRef] [Green Version]
- Okser, S.; Pahikkala, T.; Airola, A.; Salakoski, T.; Ripatti, S.; Aittokallio, T. Regularized machine learning in the genetic prediction of complex traits. PLoS Genet. 2014, 10, e1004754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wray, N.R.; Yang, J.; Hayes, B.J.; Price, A.L.; Goddard, M.E.; Visscher, P.M. Pitfalls of predicting complex traits from SNPs. Nat. Rev. Genet. 2013, 14, 507–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudbridge, F. Power and predictive accuracy of polygenic risk scores. PLoS Genet. 2013, 9, e1003348. [Google Scholar] [CrossRef]
- International Schizophrenia Consortium; Purcell, S.M.; Wray, N.R.; Stone, J.L.; Visscher, P.M.; O’Donovan, M.C.; Sullivan, P.F.; Sklar, P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009, 460, 748. [Google Scholar] [PubMed]
- Wray, R.N.; Goddard, M.E.; Visscher, P.M. Prediction of individual genetic risk to disease from genome-wide association studies. Genome Res. 2007, 17, 1520–1528. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.J.; Wedow, R.; Okbay, A.; Kong, E.; Maghzian, O.; Zacher, M.; Nguyen-Viet, T.A.; Bowers, P.; Sidorenko, J.; Karlsson Linner, R.; et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat. Genet. 2018, 50, 1112–1121. [Google Scholar] [CrossRef] [Green Version]
- Bigdeli, T.B.; Genovese, G.; Georgakopoulos, P.; Meyers, J.L.; Peterson, R.E.; Iyegbe, C.O.; Medeiros, H.; Valderrama, J.; Achtyes, E.D.; Kotov, R.; et al. Contributions of common genetic variants to risk of schizophrenia among individuals of African and Latino ancestry. Mol. Psychiatry 2020, 25, 2455–2467. [Google Scholar] [CrossRef] [Green Version]
- Rammos, A.; Gonzalez, L.A.N.; The Schizophrenia Working Group of the Psychiatric Genomics Consortium 2; Weinberger, D.R.; Mitchell, K.J.; Nicodemus, K.K. The role of polygenic risk score gene-set analysis in the context of the omnigenic model of schizophrenia. Neuropsychopharmacology 2019, 44, 1562–1569. [Google Scholar] [CrossRef]
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef]
- Qi, Q.; Stilp, A.M.; Sofer, T.; Moon, J.Y.; Hidalgo, B.; Szpiro, A.A.; Wang, T.; Ng, M.C.Y.; Guo, X.; MEta-analysis of type 2 DIabetes in African Americans (MEDIA) Consortium; et al. Genetics of Type 2 Diabetes in U.S. Hispanic/Latino Individuals: Results from the Hispanic Community Health Study/Study of Latinos (HCHS/SOL). Diabetes 2017, 66, 1419–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lall, K.; Magi, R.; Morris, A.; Metspalu, A.; Fischer, K. Personalized risk prediction for type 2 diabetes: The potential of genetic risk scores. Genet. Med. 2017, 19, 322–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, T.J.; Ehret, G.B.; Nandakumar, P.; Ranatunga, D.; Schaefer, C.; Kwok, P.Y.; Iribarren, C.; Chakravarti, A.; Risch, N. Genome-wide association analyses using electronic health records identify new loci influencing blood pressure variation. Nat. Genet. 2017, 49, 54–64. [Google Scholar] [CrossRef] [Green Version]
- Wray, N.R.; Ripke, S.; Mattheisen, M.; Trzaskowski, M.; Byrne, E.M.; Abdellaoui, A.; Adams, M.J.; Agerbo, E.; Air, T.M.; Andlauer, T.M.F.; et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 2018, 50, 668–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Yang, S.; Squirrell, D.; Vaghefi, E. Towards implementation of AI in New Zealand national diabetic screening program: Cloud-based, robust, and bespoke. PLoS ONE 2020, 15, e0225015. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.W.; Tafe, L.J.; Linnik, Y.A.; Vaickus, L.J.; Tomita, N.; Hassanpour, S. Pathologist-level classification of histologic patterns on resected lung adenocarcinoma slides with deep neural networks. Sci. Rep. 2019, 9, 3358. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Lei, C.; Huang, C.J.; Kobayashi, H.; Sun, C.W.; Goda, K. Intelligent Image De-Blurring for Imaging Flow Cytometry. Cytom. A 2019, 95, 549–554. [Google Scholar] [CrossRef]
- Chai, Y.; Liu, H.; Xu, J. Glaucoma diagnosis based on both hidden features and domain knowledge through deep learning models. Knowl. Based Syst. 2018, 161, 147–156. [Google Scholar] [CrossRef]
- Jeyaraj, R.P.; Nadar, E.R.S. Computer-assisted medical image classification for early diagnosis of oral cancer employing deep learning algorithm. J. Cancer Res. Clin. Oncol. 2019, 145, 829–837. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sonderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; Von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Rahman, R.; Kodesh, A.; Levine, S.Z.; Sandin, S.; Reichenberg, A.; Schlessinger, A. Identification of newborns at risk for autism using electronic medical records and machine learning. Eur. Psychiatry 2020, 63, e22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamate, D.; Kim, M.; Proitsi, P.; Westwood, S.; Baird, A.; Nevado-Holgado, A.; Hye, A.; Bos, I.; Vos, S.J.B.; Vandenberghe, R.; et al. A metabolite-based machine learning approach to diagnose Alzheimer-type dementia in blood: Results from the European Medical Information Framework for Alzheimer disease biomarker discovery cohort. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Hennig, H.; Rees, P.; Blasi, T.; Kamentsky, L.; Hung, J.; Dao, D.; Carpenter, A.E.; Filby, A. An open-source solution for advanced imaging flow cytometry data analysis using machine learning. Methods 2017, 112, 201–210. [Google Scholar] [CrossRef] [Green Version]
- Elaziz, M.A.; Hosny, K.M.; Salah, A.; Darwish, M.M.; Lu, S.T.; Sahlol, A.T. New machine learning method for image-based diagnosis of COVID-19. PLoS ONE 2020, 15, e0235187. [Google Scholar] [CrossRef]
- Mir, A.; Dhage, S.N. Diabetes Disease Prediction Using Machine Learning on Big Data of Healthcare. In Proceedings of the 2018 Fourth International Conference on Computing Communication Control and Automation (ICCUBEA), Pune, India, 16–18 August 2018. [Google Scholar]
- Polygenic Risk Score Task Force of the International Common Disease Alliance; Adeyemo, A.; Balaconis, M.K.; Darnes, D.R.; Rippati, S.; Widen, E.; Zhou, A. Responsible use of polygenic risk scores in the clinic: Potential benefits, risks and gaps. Nat. Med. 2021, 27, 1876–1884. [Google Scholar]
- Polasek, T.M.; Tucker, G.T.; Sorich, M.J.; Wiese, M.D.; Mohan, T.; Rostami-Hodjegan, A.; Korprasertthaworn, P.; Perera, V.; Rowland, A. Prediction of olanzapine exposure in individual patients using physiologically based pharmacokinetic modelling and simulation. Br. J. Clin. Pharmacol. 2018, 84, 462–476. [Google Scholar] [CrossRef] [Green Version]
- Lippert, J.; Brosch, M.; Von Kampen, O.; Meyer, M.; Siegmund, H.U.; Schafmayer, C.; Becker, T.; Laffert, B.; Gorlitz, L.; Schreiber, S.; et al. A mechanistic, model-based approach to safety assessment in clinical development. CPT Pharmacomet. Syst. Pharm. 2012, 1, e13. [Google Scholar] [CrossRef]
- Rasool, M.F.; Ali, S.; Khalid, S.; Khalid, R.; Majeed, A.; Imran, I.; Saeed, H.; Usman, M.; Ali, M.; Alali, A.S.; et al. Development and evaluation of physiologically based pharmacokinetic drug-disease models for predicting captopril pharmacokinetics in chronic diseases. Sci. Rep. 2021, 11, 8589. [Google Scholar] [CrossRef]
- Heimbach, T.; Chen, Y.; Chen, J.; Dixit, V.; Parrott, N.; Peters, S.A.; Poggesi, I.; Sharma, P.; Snoeys, J.; Shebley, M.; et al. Physiologically-Based Pharmacokinetic Modeling in Renal and Hepatic Impairment Populations: A Pharmaceutical Industry Perspective. Clin. Pharmacol. Ther. 2021, 110, 297–310. [Google Scholar] [CrossRef]
- Shah, K.; Fischetti, B.; Cha, A.; Taft, D.R. Using PBPK Modeling to Predict Drug Exposure and Support. Dosage Adjustments in Patients with Renal Impairment: An Example with Lamivudine. Curr. Drug Discov. Technol. 2020, 17, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Fendt, R.; Hofmann, U.; Schneider, A.R.P.; Schaeffeler, E.; Burghaus, R.; Yilmaz, A.; Blank, L.M.; Kerb, R.; Lippert, J.; Schlender, J.F.; et al. Data-driven personalization of a physiologically based pharmacokinetic model for caffeine: A systematic assessment. CPT Pharmacomet. Syst. Pharm. 2021, 10, 782–793. [Google Scholar] [CrossRef]
- Debray, T.P.; Riley, R.D.; Rovers, M.M.; Reitsma, J.B.; Moons, K.G.; Cochrane IPD Meta-analysis Methods group. Individual participant data (IPD) meta-analyses of diagnostic and prognostic modeling studies: Guidance on their use. PLoS Med. 2015, 12, e1001886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldi, P.; Brunak, S.; Chauvin, Y.; Andersen, C.; Nielsen, H. Assessing the accuracy of prediction algorithms for classification: An overview. Bioinformatics 2000, 16, 412–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharm, M.; Waltemath, D. A fully featured COMBINE archive of a simulation study on syncytial mitotic cycles in Drosophila embryos. F1000Research 2016, 5, 2421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golebiewski, M. Data Formats for Systems Biology and Quantitative Modeling. In Encyclopedia of Bioinformatics and Computational Biology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 884–893. [Google Scholar]
- Cohen, J.F.; Korevaar, D.A.; Altman, D.G.; Bruns, D.E.; Gatsonis, C.A.; Hooft, L.; Irwig, L.; Levine, D.; Reitsma, J.B.; De Vet, H.C.; et al. STARD 2015 guidelines for reporting diagnostic accuracy studies: Explanation and elaboration. BMJ Open 2016, 6, e012799. [Google Scholar] [CrossRef] [PubMed]
- Enderling, H.; Wolkenhauer, O. Are all models wrong? Comput. Syst. Oncol. 2020, 1, e1008. [Google Scholar] [CrossRef]



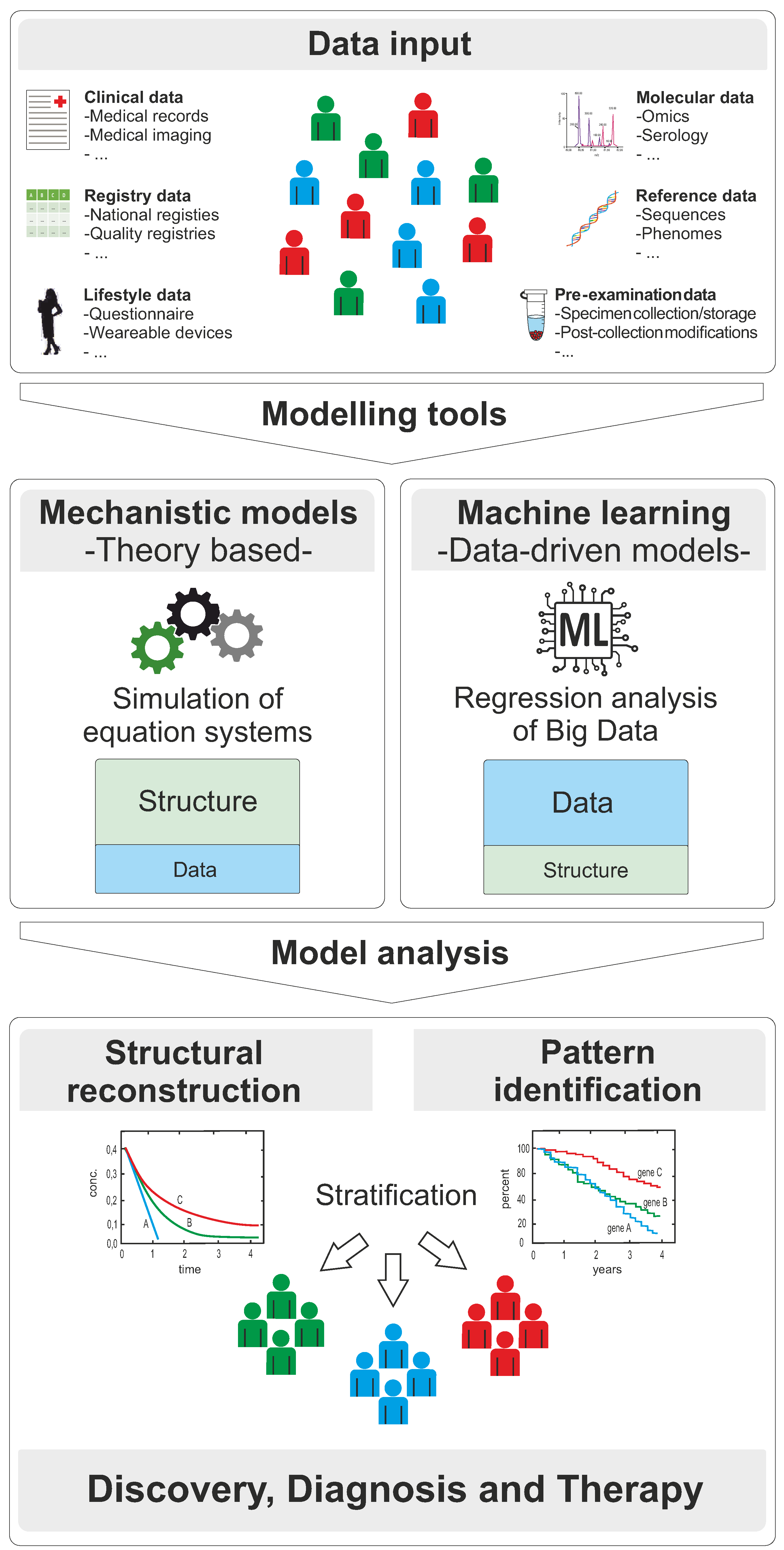{kind=link}
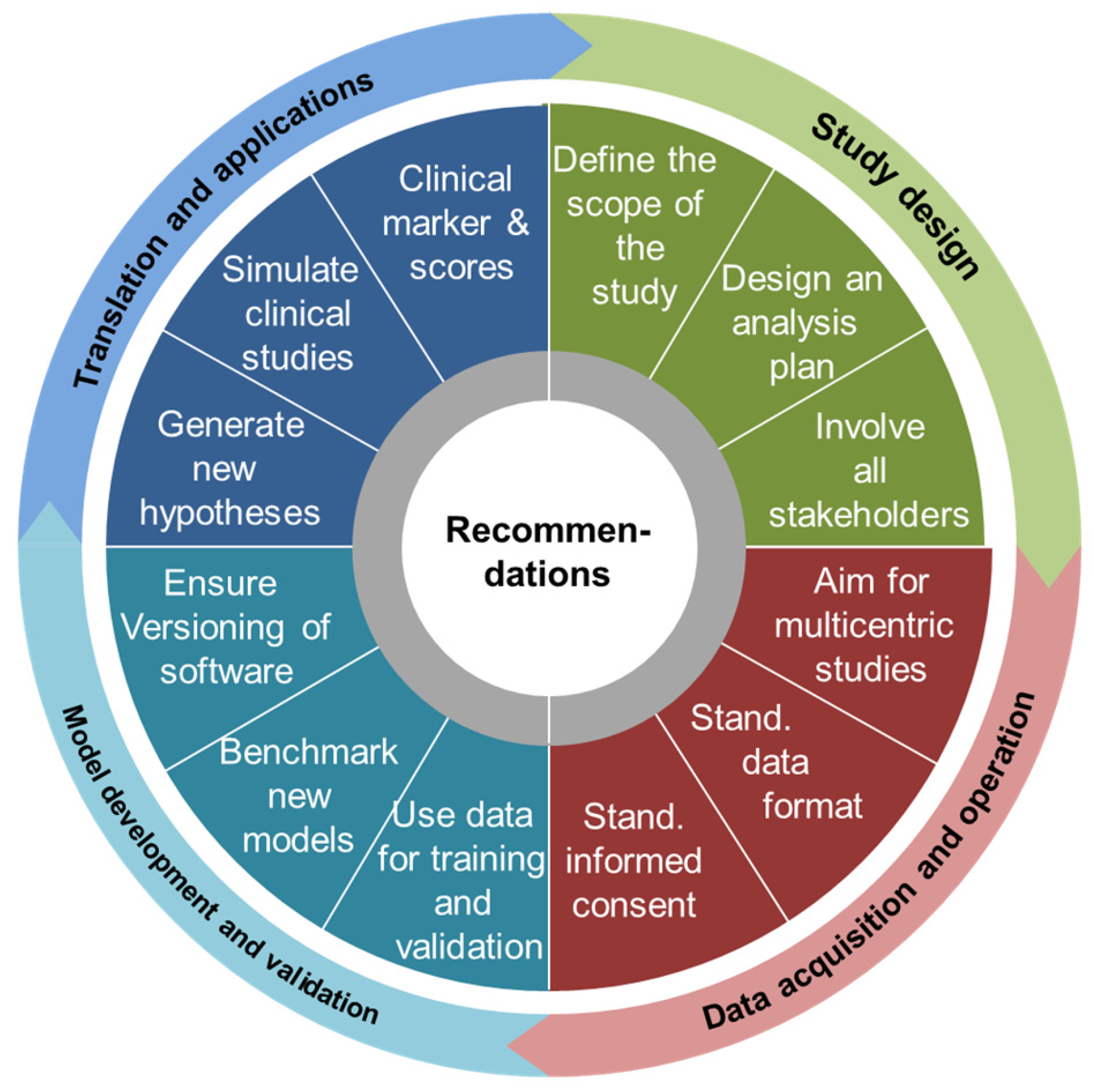{kind=link}
| Research Field | Resources | Tools |
|---|---|---|
| Molecular interaction maps | SIGnaling Network Open Resource (SIGNOR) [40], Reactome [41], SignaLink [42], InnateDB [43], Atlas of Cancer Signalling Network (ACSN) [15], OmniPath [35], RING [36], WikiPathways [44], Kyoto Encyclopedia of Genes and Genomes (KEGG) [45] | CellDesigner [46], Cytoscape & plugins [47], Molecular Interaction NEtwoRks VisuAlization (MINERVA) [48], NaviCell [49], Newt [50] |
| Boolean models | CellNetAnalyzer (CNA) [37], CellCollective [38], GINsim [39], PyBoolNet repository [46], BioModels [47] | CNA [37], Genetic Network Analyzer (GNA) [48], CellCollective [38], GINsim [39], SQUAD-Boolsim [49], BoolNet [18], Markovian Boolean Stochastic Simulator (MaBoSS) [50], CellNOpt [51] |
| Constrained-based models | BioModels [47], BiGG (Biochemical, Genetic and Genomic knowledge base) [52], Human metabolic atlas [53], Virtual Metabolic Human [54] | COnstraint-based Reconstruction and Analysis (COBRA) toolbox [55], Sybil package [56], COBRApy [57], ModelSEED [58] |
| Quantitative models | BioModels [59], Java Web Simulation (JWS) [60], Physiome Model Repository [61] | COmplex PAthway Simulator (COPASI) [62], CellDesigner [63], JWS [60] |
| Pharmacokinetic models | PharmML (Pharmacometrics Markup Language [30], Open Systems Pharmacology | Monolix, SimCypTM, GastroPlus®, PK-Sim® |
| Research Field | Content |
|---|---|
| Molecular interaction maps | |
| Inflammation | Knowledge-base, disease mechanisms, data interpretation [83] |
| Neurodegenerative disease | Knowledge-base, disease mechanisms, data interpretation [14] |
| Cancer | Knowledge-base, disease mechanisms, data interpretation [15] |
| Rheumatoid Arthritis | Knowledge-base, critical nodes (drug targets) [84] |
| Asthma | Disease mechanisms [85] |
| Atherosclerosis | Disease mechanisms, data interpretation, critical nodes (drug targets) [86] |
| Boolean models | |
| Cancer | Disease mechanism, patient stratification [17] |
| Type 2 diabetes | Disease mechanism, patient stratification [87] |
| Obesity | Disease mechanism, patient stratification [18] |
| Non-alcoholic fatty liver disease | Disease mechanism, patient stratification [88] |
| Genome-scale metabolic models | |
| Cancer | Disease markers, drug targets, patient stratification [22] |
| Auto-Immune diseases | Target identification, biomarkers, patient stratification [89] |
| Cancer | Personalized combination therapy [21] |
| Cancer | Disease signature, drug targets, patient stratification [90] |
| Cancer | Disease markers, drug targets, patient stratification [22] |
| Auto-Immune diseases | Target identification, biomarkers, patient stratification [89] |
| Cancer | Personalized combination therapy [21] |
| Research Field | Content |
|---|---|
| Deep Learning and Convolutional Neural Network Models | |
| Ophthalmology | The first FDA-authorized autonomous AI system for the detection of diabetic retinopathy [100] |
| Radiology | DL based model that is able to detect COVID-19-induced pneumonia on chest X-ray images [101] |
| Ophthalmology | Two models for quality assurance and diagnosis of diabetic retinopathy on retinal images [121] |
| Pathology | Assistance to pathologists for improving classification of lung adenocarcinoma patterns by automatically pre-screening and highlighting cancerous regions prior to review [122] |
| Imaging flow cytometry | Automated image de-blurring of out-of-focus cells in imaging flow cytometry [123] |
| Ophthalmology | A DL model for the diagnosis of glaucoma based upon images and domain knowledge features [124] |
| Oncology | Automated detection of oral cancer on hyperspectral images [125] |
| Deep Learning and Deconvolutional Neural Network Models | |
| Proteomics | Neural network that is able to predict signal peptides (SP) from amino-acid sequences and distinguish between three groups of prokaryotic SPs [126] |
| Antibody engineering | Prediction of antigen specificity via DL, which leads to optimized antibody variants for therapeutic purposes [96] |
| Intensive care | ML analysis of time-series data in intensive care units led to an improvement in the prediction of 90-day mortality [71] |
| Deep Learning, Machine Learning, Random Forest, and Deconvolutional Neural Network Models | |
| Psychiatry | A model that detects autism spectrum disorder risk for newborns with up to 95.62% from electronic medical records [127] |
| Neurology | A study with the aim to differentiate between cognitive normal people and patients with Alzheimer’s disease using various ML/DL techniques on blood metabolite levels [128] |
| Machine Learning and Polygenic Risk Score Models | |
| Coronary artery disease | Patients with high genome-wide PRS for coronary artery disease may receive greater clinical benefit from alirocumab treatment in the ODYSSEY OUTCOMES trial [102] |
| Coronary artery disease, atrial fibrillation, type 2 diabetes, inflammatory bowel disease, and breast cancer | Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Use of PRS to identify individuals at high risk for a given disease to enable enhanced screening or preventive therapies [116] |
| Machine Learning, Self-Organizing Maps, Random Forest, K-Nearest Neighbors, Support Vector Machines, Self-Operating Maps | |
| Metabolomics | SOM analysis of response metabolites detected by mass-spectroscopy leads to the identification of similar responses (ML/Self, Organizing Maps (SOM)) [97] |
| Imaging flow cytometry | An open-source toolbox for the analysis of imaging flow cytometry images (ML/RF) [129] |
| Radiology | Classification of COVID-19 and non-COVID-19 patients based on features extracted from chest X-ray images (ML/KNN) [130] |
| Endocrinology | Prediction of diabetes based on several blood values and other patient indices (ML/SVM, RF) [131] |
| Metabolomics | SOM analysis of response metabolites detected by mass-spectroscopy leads to the identification of similar responses (ML/SOM)) [97] |
| Research Field | Content |
|---|---|
| Mechanistic Models | |
| Pediatrics | Pediatric extrapolation [28] |
| Geriatrics | Geriatric extrapolation [33] |
| MIPD | Prediction of personalized drug exposure [133] |
| Pharmaco-genomics | Prediction of the incidence rates of myopathy in different genotypes [134] |
| Disease models | Prediction of drug PK in cirrhotic patients [135] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Collin, C.B.; Gebhardt, T.; Golebiewski, M.; Karaderi, T.; Hillemanns, M.; Khan, F.M.; Salehzadeh-Yazdi, A.; Kirschner, M.; Krobitsch, S.; EU-STANDS4PM consortium; et al. Computational Models for Clinical Applications in Personalized Medicine—Guidelines and Recommendations for Data Integration and Model Validation. J. Pers. Med. 2022, 12, 166. https://doi.org/10.3390/jpm12020166
Collin CB, Gebhardt T, Golebiewski M, Karaderi T, Hillemanns M, Khan FM, Salehzadeh-Yazdi A, Kirschner M, Krobitsch S, EU-STANDS4PM consortium, et al. Computational Models for Clinical Applications in Personalized Medicine—Guidelines and Recommendations for Data Integration and Model Validation. Journal of Personalized Medicine. 2022; 12(2):166. https://doi.org/10.3390/jpm12020166
Chicago/Turabian StyleCollin, Catherine Bjerre, Tom Gebhardt, Martin Golebiewski, Tugce Karaderi, Maximilian Hillemanns, Faiz Muhammad Khan, Ali Salehzadeh-Yazdi, Marc Kirschner, Sylvia Krobitsch, EU-STANDS4PM consortium, and et al. 2022. "Computational Models for Clinical Applications in Personalized Medicine—Guidelines and Recommendations for Data Integration and Model Validation" Journal of Personalized Medicine 12, no. 2: 166. https://doi.org/10.3390/jpm12020166