
DNA Methylation Analysis Reveals Distinct Patterns in Satellite Cell–Derived Myogenic Progenitor Cells of Subjects with Spastic Cerebral Palsy
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subject Enrollment
2.2. Satellite Cell Isolation
2.3. DNA Extraction, Library Preparation, and Sequencing
2.4. Methylation Analysis
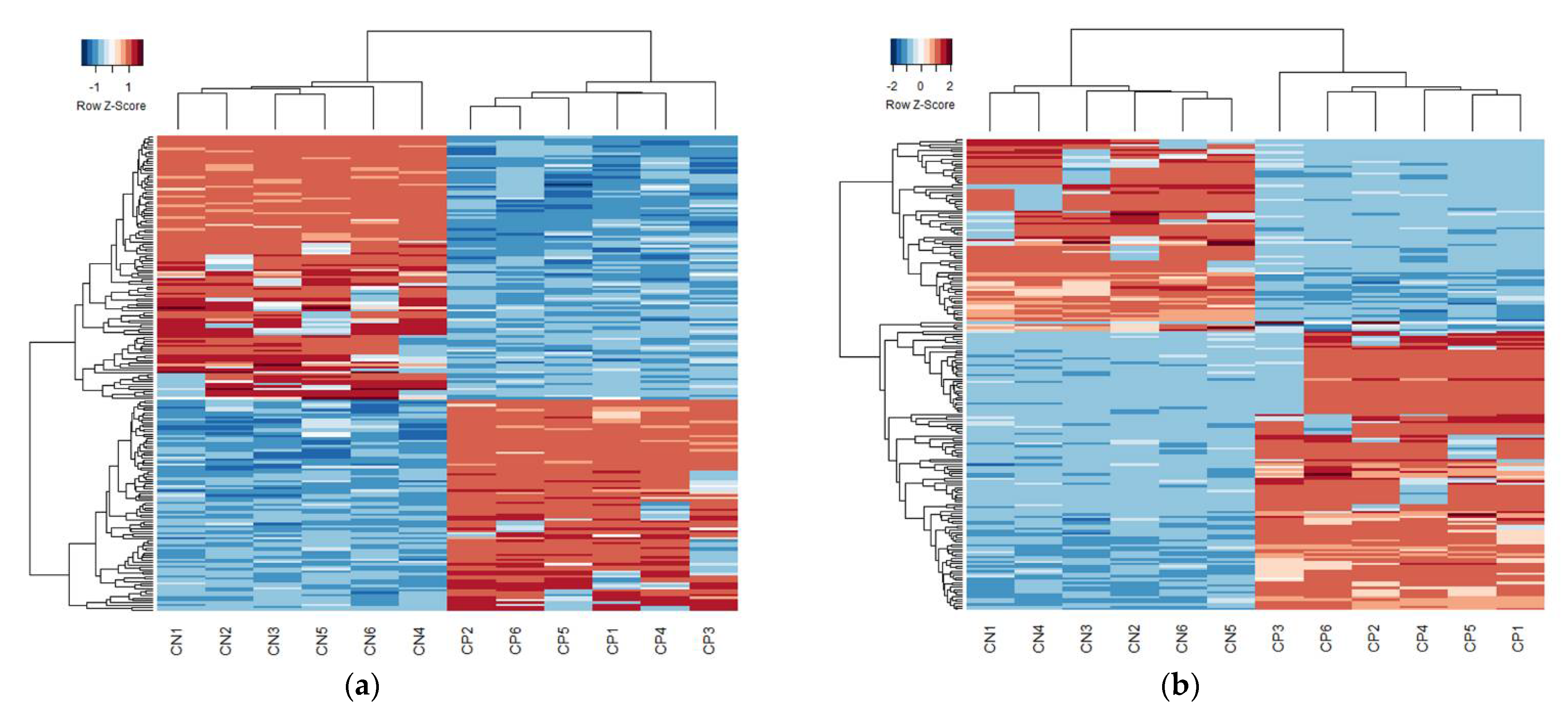
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Christensen, D.; Braun, K.V.N.; Doernberg, N.S.; Maenner, M.J.; Arneson, C.L.; Durkin, M.S.; Benedict, R.E.; Kirby, R.S.; Wingate, M.S.; Fitzgerald, R.; et al. Prevalence of cerebral palsy, co–occurring autism spectrum disorders, and motor functioning—Autism and Developmental Disabilities Monitoring Network, USA, 2008. Dev. Med. Child Neurol. 2013, 56, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, H.K.; Rosenbaum, P.; Paneth, N.; Dan, B.; Lin, J.P.; Damiano, D.L.; Becher, J.G.; Gaebler–Spira, D.; Colver, A.; Lieber, R.L.; et al. Cerebral palsy. Nat. Rev. Dis. Prim. 2016, 2, 15082. [Google Scholar] [CrossRef] [PubMed]
- Mandaleson, A.; Lee, Y.; Kerr, C.; Graham, H.K. Classifying cerebral palsy: Are we nearly there? J. Pediatr. Orthop. 2015, 35, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Oskoui, M.; Coutinho, F.; Dykeman, J.; Jetté, N.; Pringsheim, T. An update on the prevalence of cerebral palsy: A systematic review and meta–analysis. Dev. Med. Child Neurol. 2013, 55, 509–519. [Google Scholar] [CrossRef]
- NICHD. What Are the Types of Cerebral Palsy? Available online: https://www.nichd.nih.gov/health/topics/cerebral–palsy/conditioninfo/types (accessed on 14 April 2022).
- CDC. Data and Statistics for Cerebral Palsy. Available online: https://www.cdc.gov/ncbddd/cp/data.html (accessed on 14 April 2022).
- Accardo, P. Neurodevelopmental Disabilities in Infancy and Childhood, 3rd ed.; Volume I: Neurodevelopmental Diagnosis and Treatment; Paul H. Brookes Publishing Co.: Baltimore, MD, USA, 2007. [Google Scholar]
- Lieber, R.L.; Friden, J. Spasticity causes a fundamental rearrangement of muscle–joint interaction. Muscle Nerve 2002, 25, 265–270. [Google Scholar] [CrossRef]
- Maenner, M.J.; Blumberg, S.J.; Kogan, M.D.; Christensen, D.; Yeargin–Allsopp, M.; Schieve, L.A. Prevalence of cerebral palsy and intellectual disability among children identified in two U.S. National Surveys, 2011–2013. Ann. Epidemiol. 2016, 26, 222–226. [Google Scholar] [CrossRef] [Green Version]
- Mockford, M.; Caulton, J.M. The Pathophysiological Basis of Weakness in Children With Cerebral Palsy. Pediatr. Phys. Ther. 2010, 22, 222–233. [Google Scholar] [CrossRef]
- Sankar, C.; Mundkur, N. Cerebral palsy–definition, classification, etiology and early diagnosis. Indian J. Pediatr. 2005, 72, 865–868. [Google Scholar] [CrossRef]
- Sharan, D. Cerebral Palsy—Challenges for the Future; InTech: Vienna, Austria, 2014. [Google Scholar]
- Von Walden, F.; Gantelius, S.; Liu, C.; Borgström, H.; Björk, L.; Gremark, O.; Stål, P.; Nader, G.A.; Pontén, E. Muscle contractures in patients with cerebral palsy and acquired brain injury are associated with extracellular matrix expansion, pro–inflammatory gene expression, and reduced rRNA synthesis. Muscle Nerve 2018, 58, 277–285. [Google Scholar] [CrossRef]
- Oberhofer, K.; Stott, N.; Mithraratne, K.; Anderson, I. Subject–specific modelling of lower limb muscles in children with cerebral palsy. Clin. Biomech. 2010, 25, 88–94. [Google Scholar] [CrossRef]
- Bandholm, T.; Magnusson, P.; Jensen, B.R.; Sonne–Holm, S. Dorsiflexor muscle–group thickness in children with cerebral palsy: Relation to cross–sectional area. NeuroRehabilitation 2009, 24, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Fry, N.R.; Gough, M.; McNee, A.E.; Shortland, A.P. Changes in the Volume and Length of the Medial Gastrocnemius After Surgical Recession in Children With Spastic Diplegic Cerebral Palsy. J. Pediatr. Orthop. 2007, 27, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Lampe, R.; Grassl, S.; Mitternacht, J.; Gerdesmeyer, L.; Gradinger, R. MRT–measurements of muscle volumes of the lower extremities of youths with spastic hemiplegia caused by cerebral palsy. Brain Dev. 2006, 28, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Barber, L.; Hastings–Ison, T.; Baker, R.; Barrett, R.; Lichtwark, G. Medial gastrocnemius muscle volume and fascicle length in children aged 2 to 5 years with cerebral palsy. Dev. Med. Child Neurol. 2011, 53, 543–548. [Google Scholar] [CrossRef]
- de Bruin, M.; Smeulders, M.J.; Kreulen, M. Why is joint range of motion limited in patients with cerebral palsy? J. Hand Surg. (Eur. Vol.) 2013, 38, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Elder, G.C.B.; Kirk, J.; Stewart, G.; Cook, K.; Weir, D.; Marshall, A.; Leahey, L. Contributing factors to muscle weakness in children with cerebral palsy. Dev. Med. Child Neurol. 2003, 45, 542–550. [Google Scholar] [CrossRef]
- Hussain, A.W.; Onambélé, G.L.; Williams, A.G.; Morse, C.I. Muscle size, activation, and coactivation in adults with cerebral palsy. Muscle Nerve 2013, 49, 76–83. [Google Scholar] [CrossRef]
- Smith, L.R.; Lee, K.S.; Ward, S.R.; Chambers, H.G.; Lieber, R.L. Hamstring contractures in children with spastic cerebral palsy result from a stiffer extracellular matrix and increased in vivo sarcomere length. J. Physiol. 2011, 589 Pt 10, 2625–2639. [Google Scholar] [CrossRef]
- Robinson, K.G.; Mendonca, J.L.; Militar, J.L.; Theroux, M.C.; Dabney, K.W.; Shah, S.A.; Miller, F.; Akins, R.E. Disruption of Basal Lamina Components in Neuromotor Synapses of Children with Spastic Quadriplegic Cerebral Palsy. PLoS ONE 2013, 8, e70288. [Google Scholar] [CrossRef]
- Theroux, M.C.; Akins, R.E.; Barone, C.; Boyce, B.; Miller, F.; Dabney, K.W. Neuromuscular junctions in cerebral palsy: Presence of extrajunctional acetylcholine receptors. Anesthesiology 2002, 96, 330–335. [Google Scholar] [CrossRef]
- Theroux, M.C.; Oberman, K.G.; Lahaye, J.; Boyce, B.A.; DuHadaway, D.; Miller, F.; Akins, R.E. Dysmorphic neuromuscular junctions associated with motor ability in cerebral palsy. Muscle Nerve 2005, 32, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.G.; Crowgey, E.L.; Lee, S.K.; Akins, R.E. Transcriptional analysis of muscle tissue and isolated satellite cells in spastic cerebral palsy. Dev. Med. Child Neurol. 2021, 63, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.R.; Pontén, E.; Hedström, Y.; Ward, S.R.; Chambers, H.G.; Subramaniam, S.; Lieber, R.L. Novel transcriptional profile in wrist muscles from cerebral palsy patients. BMC Med. Genom. 2009, 2, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domenighetti, A.A.; Mathewson, M.A.; Pichika, R.; Sibley, L.A.; Zhao, L.; Chambers, H.G.; Lieber, R.L. Loss of myogenic potential and fusion capacity of muscle stem cells isolated from contractured muscle in children with cerebral palsy. Am. J. Physiol. Physiol. 2018, 315, C247–C257. [Google Scholar] [CrossRef] [Green Version]
- Dayanidhi, S.; Dykstra, P.B.; Lyubasyuk, V.; McKay, B.R.; Chambers, H.G.; Lieber, R.L. Reduced satellite cell number in situ in muscular contractures from children with cerebral palsy. J. Orthop. Res. 2015, 33, 1039–1045. [Google Scholar] [CrossRef]
- Smith, L.R.; Chambers, H.G.; Subramaniam, S.; Lieber, R.L. Transcriptional Abnormalities of Hamstring Muscle Contractures in Children with Cerebral Palsy. PLoS ONE 2012, 7, e40686. [Google Scholar] [CrossRef]
- Corvelyn, M.; De Beukelaer, N.; Duelen, R.; Deschrevel, J.; Van Campenhout, A.; Prinsen, S.; Gayan-Ramirez, G.; Maes, K.; Weide, G.; Desloovere, K.; et al. Muscle Microbiopsy to Delineate Stem Cell Involvement in Young Patients: A Novel Approach for Children With Cerebral Palsy. Front. Physiol. 2020, 11, 945. [Google Scholar] [CrossRef]
- Sibley, L.A.; Broda, N.; Gross, W.R.; Menezes, A.F.; Embry, R.B.; Swaroop, V.T.; Chambers, H.G.; Schipma, M.J.; Lieber, R.L.; Domenighetti, A.A. Differential DNA methylation and transcriptional signatures characterize impairment of muscle stem cells in pediatric human muscle contractures after brain injury. FASEB J. 2021, 35, e21928. [Google Scholar] [CrossRef]
- Fahey, M.C.; Maclennan, A.H.; Kretzschmar, D.; Gecz, J.; Kruer, M.C. The genetic basis of cerebral palsy. Dev. Med. Child Neurol. 2017, 59, 462–469. [Google Scholar] [CrossRef] [Green Version]
- McMichael, G.L.; Bainbridge, M.N.; Haan, E.; Corbett, M.; Gardner, A.; Thompson, S.E.; Van Bon, B.W.M.; van Eyk, C.; Broadbent, J.; Reynolds, C.A.; et al. Whole–exome sequencing points to considerable genetic heterogeneity of cerebral palsy. Mol. Psychiatry 2015, 20, 176–182. [Google Scholar] [CrossRef]
- Kubota, N.; Yokoyama, T.; Hoshi, N.; Suyama, M. Identification of a candidate enhancer for DMRT3 involved in spastic cerebral palsy pathogenesis. Biochem. Biophys. Res. Commun. 2018, 496, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Gulati, S.; Sondhi, V. Cerebral Palsy: An Overview. Indian J. Pediatr. 2017, 85, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- Sewell, M.D.; Eastwood, D.M.; Wimalasundera, N. Managing common symptoms of cerebral palsy in children. BMJ 2014, 349, g5474. [Google Scholar] [CrossRef] [PubMed]
- Bahado–Singh, R.O.; Vishweswaraiah, S.; Aydas, B.; Mishra, N.K.; Guda, C.; Radhakrishna, U. Deep Learning/Artificial Intelligence and Blood–Based DNA Epigenomic Prediction of Cerebral Palsy. Int. J. Mol. Sci. 2019, 20, 2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowgey, E.L.; Marsh, A.G.; Robinson, K.G.; Yeager, S.K.; Akins, R.E. Epigenetic machine learning: Utilizing DNA methylation patterns to predict spastic cerebral palsy. BMC Bioinform. 2018, 19, 225. [Google Scholar] [CrossRef]
- Mohandas, N.; Bass-Stringer, S.; Maksimovic, J.; Crompton, K.; Loke, Y.J.; Walstab, J.; Reid, S.M.; Amor, D.J.; Reddihough, D.; Craig, J.M.; et al. Epigenome–wide analysis in newborn blood spots from monozygotic twins discordant for cerebral palsy reveals consistent regional differences in DNA methylation. Clin. Epigenetics 2018, 10, 25. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y. Study of global DNA methylation in monozygotic twins with cerebral palsy. Pak. J. Pharm. Sci. 2017, 30 (Suppl. S4), 1467–1473. [Google Scholar]
- Alag, A. Machine learning approach yields epigenetic biomarkers of food allergy: A novel 13–gene signature to diagnose clinical reactivity. PLoS ONE 2019, 14, e0218253. [Google Scholar] [CrossRef] [Green Version]
- Aref-Eshghi, E.; Rodenhiser, D.I.; Schenkel, L.C.; Lin, H.; Skinner, C.; Ainsworth, P.; Paré, G.; Hood, R.L.; Bulman, D.E.; Kernohan, K.D.; et al. Genomic DNA Methylation Signatures Enable Concurrent Diagnosis and Clinical Genetic Variant Classification in Neurodevelopmental Syndromes. Am. J. Hum. Genet. 2018, 102, 156–174. [Google Scholar] [CrossRef] [Green Version]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation–based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef] [Green Version]
- De Bellis, M.; Camerino, D.C.; Desaphy, J.-F. Toward precision medicine in myotonic syndromes. Oncotarget 2017, 8, 14279–14280. [Google Scholar] [CrossRef] [PubMed]
- Kulis, M.; Heath, S.; Bibikova, M.; Queirós, A.C.; Navarro, A.; Clot, G.; Martínez-Trillos, A.; Castellano, G.; Brun-Heath, I.; Pinyol, M.; et al. Epigenomic analysis detects widespread gene–body DNA hypomethylation in chronic lymphocytic leukemia. Nat. Genet. 2012, 44, 1236–1242. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, Y.; Pan, X.; Li, M.; Yang, S.; Li, S.C. DNA methylation markers for Pan–Cancer prediction by deep learning. Genes 2019, 10, 778. [Google Scholar] [CrossRef] [Green Version]
- Orozco, J.I.J.; Knijnenburg, T.A.; Manughian–Peter, A.O.; Salomon, M.P.; Barkhoudarian, G.; Jalas, J.R.; Wilmott, J.S.; Hothi, P.; Wang, X.; Takasumi, Y.; et al. Epigenetic profiling for the molecular classification of metastatic brain tumors. Nat. Commun. 2018, 9, 4627. [Google Scholar] [CrossRef]
- Queiros, A.; Villamor, N.; Clot, G.; Martineztrillos, A.; Kulis, M.; Navarro, A.; Penas, E.M.M.; Jayne, S.; Majid, A.M.S.A.; Richter, J.A.; et al. A B–cell epigenetic signature defines three biologic subgroups of chronic lymphocytic leukemia with clinical impact. Leukemia 2014, 29, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Wu, Y.-M.; Lonigro, R.J.; Vats, P.; Cobain, E.; Everett, J.; Cao, X.; Rabban, E.; Kumar-Sinha, C.; Raymond, V.; et al. Integrative clinical genomics of metastatic cancer. Nature 2017, 548, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hu, Y.; Aouizerat, B.E.; Peng, G.; Marconi, V.C.; Corley, M.J.; Hulgan, T.; Bryant, K.J.; Zhao, H.; Krystal, J.H.; et al. Machine learning selected smoking–associated DNA methylation signatures that predict HIV prognosis and mortality. Clin. Epigenetics 2018, 10, 155. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA hypermethylation in disease: Mechanisms and clinical relevance. Epigenetics 2019, 14, 1141–1163. [Google Scholar] [CrossRef] [Green Version]
- Bareja, A.; Holt, J.A.; Luo, G.; Chang, C.; Lin, J.; Hinken, A.C.; Freudenberg, J.; Kraus, W.E.; Evans, W.J.; Billin, A.N. Human and Mouse Skeletal Muscle Stem Cells: Convergent and Divergent Mechanisms of Myogenesis. PLoS ONE 2014, 9, e90398. [Google Scholar] [CrossRef] [Green Version]
- Garcia, S.M.; Tamaki, S.; Lee, S.; Wong, A.; Jose, A.; Dreux, J.; Kouklis, G.; Sbitany, H.; Seth, R.; Knott, P.D.; et al. High–Yield Purification, Preservation, and Serial Transplantation of Human Satellite Cells. Stem Cell Rep. 2018, 10, 1160–1174. [Google Scholar] [CrossRef] [Green Version]
- Marsh, A.G.; Pasqualone, A.A. DNA methylation and temperature stress in an Antarctic polychaete, Spiophanes tcherniai. Front. Physiol. 2014, 5, 173. [Google Scholar] [CrossRef] [PubMed]
- Rambo, I.M.; Marsh, A.; Biddle, J.F. Cytosine Methylation within Marine Sediment Microbial Communities: Potential Epigenetic Adaptation to the Environment. Front. Microbiol. 2019, 10, 1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Toward better understanding of artifacts in variant calling from high–coverage samples. Bioinformatics 2014, 30, 2843–2851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA–Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Belinda, P.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA–sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Li, R.; Yang, Y.-E.; Yin, Y.-H.; Zhang, M.-Y.; Li, H.; Qu, Y.-Q. Methylation and transcriptome analysis reveal lung adenocarcinoma–specific diagnostic biomarkers. J. Transl. Med. 2019, 17, 324. [Google Scholar] [CrossRef] [Green Version]
- Maksimovic, J.; Phipson, B.; Oshlack, A. A cross–package Bioconductor workflow for analysing methylation array data. F1000Research 2016, 5, 1281. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The Epigenomics of Cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [Green Version]
- Contreras-Romero, C.; Pérez-Yépez, E.-A.; Martinez-Gutierrez, A.D.; Campos-Parra, A.; Zentella-Dehesa, A.; Jacobo-Herrera, N.; López-Camarillo, C.; Corredor-Alonso, G.; Martínez-Coronel, J.; Rodríguez-Dorantes, M.; et al. Gene Promoter–Methylation Signature as Biomarker to Predict Cisplatin–Radiotherapy Sensitivity in Locally Advanced Cervical Cancer. Front. Oncol. 2022, 12, 773438. [Google Scholar] [CrossRef]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Flicek, P.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef] [PubMed]
- University of California Santa Cruz (UCSC) Genome Browser Gateway. Available online: https://genome.ucsc.edu/ (accessed on 15 November 2022). (accessed multiple times in 2021 and 2022 with final access date for validation).
- Robertson, K.D. DNA methylation and chromatin—Unraveling the tangled web. Oncogene 2002, 21, 5361–5379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaze, J.; Roth, T.L. Evidence from clinical and animal model studies of the long–term and transgenerational impact of stress on DNA methylation. Semin. Cell Dev. Biol. 2015, 43, 76–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartley, I.; Elkhoury, F.F.; Shin, J.H.; Xie, B.; Gu, X.; Gao, Y.; Zhou, D.; Haddad, G.G. Long–Lasting Changes in DNA Methylation Following Short–Term Hypoxic Exposure in Primary Hippocampal Neuronal Cultures. PLoS ONE 2013, 8, e77859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houtepen, L.C.; Vinkers, C.H.; Carrillo–Roa, T.; Hiemstra, M.; Van Lier, P.A.; Meeus, W.; Branje, S.; Heim, C.; Nemeroff, C.B.; Mill, J.; et al. Genome–wide DNA methylation levels and altered cortisol stress reactivity following childhood trauma in humans. Nat. Commun. 2016, 7, 10967. [Google Scholar] [CrossRef] [Green Version]
- Labonté, B.; Suderman, M.; Maussion, G.; Navaro, L.; Yerko, V.; Mahar, I.; Bureau, A.; Mechawar, N.; Szyf, M.; Meaney, M.J.; et al. Genome–wide Epigenetic Regulation by Early–Life Trauma. Arch. Gen. Psychiatry 2012, 69, 722–731. [Google Scholar] [CrossRef] [Green Version]
- Pacis, A.; Tailleux, L.; Morin, A.M.; Lambourne, J.; MacIsaac, J.L.; Yotova, V.; Dumaine, A.; Danckaert, A.; Luca, F.; Grenier, J.-C.; et al. Bacterial infection remodels the DNA methylation landscape of human dendritic cells. Genome Res. 2015, 25, 1801–1811. [Google Scholar] [CrossRef] [Green Version]
- Rask–Andersen, M.; Martinsson, D.; Ahsan, M.; Enroth, S.; Ek, W.E.; Gyllensten, U.; Johansson, Å. Epigenome–wide association study reveals differential DNA methylation in individuals with a history of myocardial infarction. Hum. Mol. Genet. 2016, 25, 4739–4748. [Google Scholar] [CrossRef] [Green Version]
- Stirzaker, C.; Zotenko, E.; Song, J.Z.; Qu, W.; Nair, S.S.; Locke, W.; Stone, A.; Armstong, N.J.; Robinson, M.D.; Dobrovic, A.; et al. Methylome sequencing in triple–negative breast cancer reveals distinct methylation clusters with prognostic value. Nat. Commun. 2015, 6, 5899. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.; Zhao, Y.; Jin, R.; Chen, J.; Wang, X.; Baccarelli, A.; Zhang, Y. Fetal growth restriction and methylation of growth–related genes in the placenta. Epigenomics 2016, 8, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Carrió, E.; Lois, S.; Mallona, I.; Cases, I.; Forn, M.; Peinado, M.A.; Suelves, M.; Díez–Villanueva, A. Deconstruction of DNA Methylation Patterns During Myogenesis Reveals Specific Epigenetic Events in the Establishment of the Skeletal Muscle Lineage. Stem Cells 2015, 33, 2025–2036. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.M.; Fry, R.C. Environmental Influences on the Epigenome: Exposure– Associated DNA Methylation in Human Populations. Annu. Rev. Public Health 2018, 39, 309–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosnowski, D.W.; Booth, C.; York, T.P.; Amstadter, A.B.; Kliewer, W. Maternal prenatal stress and infant DNA methylation: A systematic review. Dev. Psychobiol. 2018, 60, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Watamura, S.E.; Roth, T.L. Looking back and moving forward: Evaluating and advancing translation from animal models to human studies of early life stress and DNA methylation. Dev. Psychobiol. 2018, 61, 323–340. [Google Scholar] [CrossRef] [PubMed]
- Bustelo, M.; Barkhuizen, M.; Hove, D.L.A.V.D.; Steinbusch, H.W.M.; Bruno, M.A.; Loidl, C.F.; Gavilanes, A.W.D. Clinical Implications of Epigenetic Dysregulation in Perinatal Hypoxic–Ischemic Brain Damage. Front. Neurol. 2020, 11, 483. [Google Scholar] [CrossRef]
- Menon, R.; Conneely, K.N.; Smith, A.K. DNA Methylation: An Epigenetic Risk Factor in Preterm Birth. Reprod. Sci. 2012, 19, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Richetto, J.; Massart, R.; Weber–Stadlbauer, U.; Szyf, M.; Riva, M.A.; Meyer, U. Genome–wide DNA Methylation Changes in a Mouse Model of Infection–Mediated Neurodevelopmental Disorders. Biol. Psychiatry 2016, 81, 265–276. [Google Scholar] [CrossRef]
- Vaiserman, A.M. Epigenetic programming by early–life stress: Evidence from human populations. Dev. Dyn. 2014, 244, 254–265. [Google Scholar] [CrossRef]
- Smith, L.; Chambers, H.G.; Lieber, R.L. Reduced satellite cell population may lead to contractures in children with cerebral palsy. Dev. Med. Child Neurol. 2012, 55, 264–270. [Google Scholar] [CrossRef] [Green Version]
- Sharples, A.P.; Polydorou, I.; Hughes, D.; Owens, D.J.; Hughes, T.M.; Stewart, C. Skeletal muscle cells possess a ‘memory’ of acute early life TNF–α exposure: Role of epigenetic adaptation. Biogerontology 2015, 17, 603–617. [Google Scholar] [CrossRef]
- Tsumagari, K.; Baribault, C.; Terragni, J.; Varley, K.; Gertz, J.; Pradhan, S.; Badoo, M.; Crain, C.M.; Song, L.; Crawford, G.E.; et al. Early de novo DNA methylation and prolonged demethylation in the muscle lineage. Epigenetics 2013, 8, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, X.; Lu, C. The interplay between DNA and histone methylation: Molecular mechanisms and disease implications. EMBO Rep. 2021, 22, e51803. [Google Scholar] [CrossRef] [PubMed]
- Lea, A.J.; Vockley, C.M.; Johnston, R.A.; Del Carpio, C.A.; Barreiro, L.; Reddy, T.E.; Tung, J. Genome–wide quantification of the effects of DNA methylation on human gene regulation. eLife 2018, 7, e37513. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.R.; Busche, S.; Ge, B.; Kwan, T.; Pastinen, T.; Blanchette, M. The relationship between DNA methylation, genetic and expression inter–individual variation in untransformed human fibroblasts. Genome Biol. 2014, 15, R37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angeloni, A.; Bogdanovic, O. Enhancer DNA methylation: Implications for gene regulation. Essays Biochem. 2019, 63, 707–715. [Google Scholar] [CrossRef]
- Bae, M.G.; Kim, J.Y.; Choi, J.K. Frequent hypermethylation of orphan CpG islands with enhancer activity in cancer. BMC Med. Genom. 2016, 9 (Suppl. S1), 38. [Google Scholar] [CrossRef] [Green Version]
- Glass, J.L.; Hassane, D.; Wouters, B.J.; Kunimoto, H.; Avellino, R.; Garrett–Bakelman, F.E.; Guryanova, O.A.; Bowman, R.; Redlich, S.; Intlekofer, A.M.; et al. Epigenetic Identity in AML Depends on Disruption of Nonpromoter Regulatory Elements and Is Affected by Antagonistic Effects of Mutations in Epigenetic Modifiers. Cancer Discov. 2017, 7, 868–883. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hao, D.-P.; Li, J.-J.; Wang, L.; Di, L.-J. Genome–wide methylome and chromatin interactome identify abnormal enhancer to be risk factor of breast cancer. Oncotarget 2017, 8, 44705–44719. [Google Scholar] [CrossRef] [Green Version]
- Benton, M.L.; Talipineni, S.C.; Kostka, D.; Capra, J.A. Genome–wide enhancer annotations differ significantly in genomic distribution, evolution, and function. BMC Genom. 2019, 20, 511. [Google Scholar] [CrossRef] [Green Version]
- Romero, B.; Robinson, K.G.; Batish, M.; Akins, R.E. An Emerging Role for Epigenetics in Cerebral Palsy. J. Pers. Med. 2021, 11, 1187. [Google Scholar] [CrossRef]
- Mbadhi, M.N.; Tang, J.-M.; Zhang, J.-X. Histone Lysine Methylation and Long Non-coding RNA: The New Target Players in Skeletal Muscle Cell Regeneration. Front. Cell Dev. Biol. 2021, 9, 759237. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Lv, W.; Tong, Q.; Jin, J.; Xu, Z.; Zuo, B. Functional Non-coding RNA During Embryonic Myogenesis and Postnatal Muscle Development and Disease. Front. Cell Dev. Biol. 2021, 9, 628339. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Sample | Diagnosis | Age | Sex | GMFCS | Tissue Source |
|---|---|---|---|---|---|
| CN1 | Spondylolysis | 16.6 | M | N/A | Spinalis |
| CN2 | Torn ACL | 12.6 | M | N/A | Semitendinosus |
| CN3 | Idiopathic scoliosis | 12.1 | F | N/A | Spinalis |
| CN4 | Torn ACL | 12.7 | F | N/A | Semitendinosus |
| CN5 | Idiopathic scoliosis | 15.1 | M | N/A | Spinalis |
| CN6 | Idiopathic scoliosis | 14.3 | F | N/A | Spinalis |
| CP1 | Spastic CP | 15.6 | M | 5 | Vastus lateralis |
| CP2 | Spastic CP | 19.1 | M | 5 | Adductor longus |
| CP3 | Spastic CP | 12.6 | M | 4 | Rectus femoris |
| CP4 | Spastic CP | 13.8 | F | 2 | Rectus femoris |
| CP5 | Spastic CP | 19.0 | F | 5 | Spinalis |
| CP6 | Spastic CP | 12.8 | F | 5 | Spinalis |
| Position | MB LogFC | MB FDR Corrected p-Value | MT LogFC | MT FDR Corrected p-Value | Gene |
|---|---|---|---|---|---|
| chr2.0003882321 | 0.88 | 2.29 × 10−2 | 0.90 | 2.40 × 10−2 | |
| chr2.0029850455 | 1.24 | 6.74 × 10−4 | 1.32 | 1.11 × 10−5 | ALK |
| chr2.0033057636 | −0.84 | 3.40 × 10−3 | −0.85 | 1.94 × 10−2 | LINC00486 |
| chr2.0035092870 | −1.09 | 1.00 × 10−2 | −1.19 | 8.26 × 10−5 | AC012593.1 |
| chr2.0056193463 | 1.32 | 1.79 × 10−3 | 1.55 | 1.22 × 10−6 | RP11—481J13.1, AC011306.2 |
| chr2.0223166989 | 0.87 | 4.07 × 10−2 | 0.90 | 4.80 × 10−2 | CCDC140 |
| chr2.0235215325 | −1.05 | 6.08 × 10−4 | −1.08 | 6.49 × 10−4 | |
| chr3.0053784559 | 0.91 | 1.58 × 10−2 | 1.14 | 8.96 × 10−4 | CACNA1D |
| chr3.0060919598 | −0.85 | 1.89 × 10−3 | −0.86 | 3.09 × 10−2 | FHIT |
| chr3.0119863345 | 1.28 | 2.04 × 10−3 | 1.48 | 1.19 × 10−5 | GPR156 |
| chr3.0119990864 | −1.14 | 2.05 × 10−3 | −1.01 | 4.01 × 10−2 | GPR156 |
| chr3.0127606140 | −1.40 | 1.26 × 10−4 | −1.12 | 6.36 × 10−3 | |
| chr3.0182124231 | −1.14 | 3.82 × 10−2 | −1.15 | 5.10 × 10−3 | |
| chr3.0189791239 | −1.34 | 1.56 × 10−2 | −1.13 | 5.00 × 10−2 | LEPREL1 |
| chr3.0196595774 | −1.37 | 3.54 × 10−2 | −1.33 | 1.44 × 10−2 | SENP5 |
| chr4.0101719592 | −1.04 | 4.74 × 10−5 | −1.15 | 1.77 × 10−7 | EMCN |
| chr5.0011534641 | 0.90 | 2.48 × 10−2 | 1.05 | 7.82 × 10−3 | CTNND2 |
| chr5.0039219698 | 1.22 | 3.91 × 10−2 | 1.37 | 2.24 × 10−2 | FYB |
| chr5.0164483805 | −0.92 | 2.51 × 10−2 | −1.11 | 1.05 × 10−3 | CTC—340A15.2 |
| chr5.0166472226 | −1.03 | 1.03 × 10−2 | −1.16 | 4.86 × 10−4 | |
| chr6.0008948266 | 1.16 | 2.27 × 10−2 | 1.24 | 2.52 × 10−3 | |
| chr6.0016145414 | −1.17 | 2.94 × 10−4 | −1.24 | 9.54 × 10−5 | MYLIP |
| chr6.0019413218 | 0.81 | 2.95 × 10−3 | 0.86 | 1.70 × 10−2 | |
| chr6.0031008851 | 0.96 | 2.29 × 10−2 | 1.01 | 1.56 × 10−2 | RASSF3 |
| chr6.0154640863 | 1.37 | 9.68 × 10−3 | 1.34 | 3.64 × 10−3 | IPCEF1 |
| chr6.0161063597 | −2.29 | 3.71 × 10−3 | −1.62 | 2.31 × 10−2 | LPA |
| chr7.0016768868 | −0.75 | 1.18 × 10−2 | −1.03 | 1.03 × 10−5 | |
| chr7.0044621160 | 0.91 | 1.71 × 10−2 | 0.97 | 4.26 × 10−2 | TMED4 |
| chr7.0147581299 | −0.68 | 3.30 × 10−2 | −0.83 | 3.24 × 10−2 | CNTNAP2 |
| chr11.0123045794 | −1.36 | 1.13 × 10−3 | −1.54 | 2.08 × 10−6 | CLMP |
| chr11.0129565594 | 1.28 | 1.59 × 10−2 | 1.68 | 1.23 × 10−4 | |
| chr12.0003241735 | 1.19 | 4.26 × 10−3 | 1.04 | 3.25 × 10−2 | TSPAN9 |
| chr12.0026672531 | 1.00 | 4.67 × 10−2 | 1.28 | 1.68 × 10−2 | ITPR2 |
| chr12.0048360477 | −1.69 | 3.13 × 10−4 | −1.10 | 4.25 × 10−2 | TMEM106C |
| chr12.0054366343 | 0.87 | 4.94 × 10−2 | 1.07 | 3.53 × 10−2 | HOTAIR |
| chr12.0055783991 | 1.19 | 4.36 × 10−2 | 1.29 | 2.11 × 10−2 | |
| chr12.0083436417 | 1.62 | 4.94 × 10−3 | 2.14 | 6.72 × 10−5 | TMTC2 |
| chr12.0114887843 | 1.42 | 1.84 × 10−4 | 0.62 | 4.91 × 10−2 | |
| chr12.0116068191 | −1.32 | 1.42 × 10−2 | −1.58 | 5.07 × 10−6 | RP11—1028N23.4 |
| chr12.0128167651 | 1.13 | 3.28 × 10−2 | 1.57 | 8.87 × 1014 | |
| chr12.0131689822 | 1.29 | 2.19 × 10−2 | 1.74 | 5.09 × 10−5 | RP11—638F5.1 |
| chr13.0021286449 | −1.10 | 4.87 × 10−2 | −1.33 | 2.22 × 10−2 | IL17D |
| chr13.0027424109 | 1.51 | 1.23 × 10−2 | 1.56 | 5.47 × 10−4 | |
| chr13.0033220266 | −1.24 | 5.87 × 10−3 | −1.31 | 2.18 × 10−3 | PDS5B |
| chr13.0047191668 | −1.30 | 6.51 × 10−3 | −1.34 | 6.36 × 10−3 | LRCH1 |
| chr13.0093896533 | 1.50 | 2.96 × 10−2 | 2.41 | 2.86 × 10−5 | GPC6 |
| chr13.0099687193 | 1.01 | 3.54 × 10−2 | 1.06 | 4.85 × 10−2 | DOCK9 |
| chr13.0107176083 | −1.69 | 7.74 × 10−3 | −1.68 | 1.29 × 10−2 | EFNB2 |
| chr13.0109856377 | −1.48 | 4.00 × 10−2 | −1.52 | 6.19 × 10−3 | MYO16 |
| chr14.0021177142 | −1.22 | 3.13 × 10−4 | −1.24 | 7.38 × 10−5 | |
| chr14.0021316565 | −1.29 | 2.23 × 10−2 | −1.56 | 2.05 × 10−4 | |
| chr14.0025947530 | 0.91 | 1.48 × 10−2 | 1.05 | 7.13 × 10−3 | |
| chr14.0080449863 | −1.84 | 5.11 × 10−5 | −1.74 | 2.05 × 10−4 | |
| chr14.0085404000 | −1.38 | 4.50 × 10−3 | −1.38 | 3.07 × 10−3 | |
| chr14.0104190006 | −1.48 | 3.71 × 10−3 | −1.75 | 2.66 × 10−4 | ZFYVE21 |
| chr15.0046178808 | −0.97 | 1.71 × 10−2 | −0.70 | 4.78 × 10−2 | RP11—718O11.1 |
| chr15.0069824154 | 1.42 | 1.25 × 10−4 | 1.55 | 4.82 × 10−6 | RP11—279F6.1 |
| chr15.0092982723 | 1.45 | 3.91 × 10−5 | 1.73 | 9.39 × 10−9 | ST8SIA2 |
| chr16.0004815786 | −0.89 | 8.94 × 10−4 | −0.92 | 9.24 × 10−3 | ZNF500 |
| chr16.0077912976 | −1.23 | 8.49 × 10−3 | −1.28 | 2.22 × 10−3 | VAT1L |
| chr16.0079468883 | 1.11 | 4.82 × 10−3 | 1.28 | 3.78 × 10−5 | |
| chr17.0018941025 | −1.85 | 1.91 × 10−3 | −1.96 | 2.09 × 10−4 | GRAP |
| chr17.0019045779 | −1.55 | 7.74 × 10−3 | −1.56 | 8.05 × 10−4 | GRAPL, CTC—457L16.2 |
| chr17.0028803808 | −1.20 | 1.47 × 10−2 | −1.24 | 4.72 × 10−3 | |
| chr17.0070499160 | 1.04 | 1.42 × 10−4 | 1.06 | 1.48 × 10−3 | LINC00511 |
| chr17.0074566299 | 0.87 | 3.28 × 10−2 | 1.14 | 3.76 × 10−4 | ST6GALNAC2, RP11—666A8.9 |
| chr18.0043923940 | −1.66 | 5.63 × 10−6 | −1.73 | 7.53 × 10−7 | RNF165 |
| chr18.0045011716 | 1.10 | 2.73 × 10−2 | 1.41 | 4.57 × 10−5 | CTD—2130O13.1 |
| chr18.0047177650 | −1.15 | 3.29 × 10−4 | −0.64 | 8.52 × 10−3 | |
| chr18.0047230566 | −1.39 | 1.98 × 10−2 | −1.30 | 3.48 × 10−3 | |
| chr18.0072250823 | 1.12 | 7.11 × 10−3 | 1.05 | 2.58 × 10−2 | CNDP1 |
| chr19.0002867898 | 1.36 | 5.19 × 10−9 | 1.12 | 1.72 × 10−2 | ZNF556 |
| chr19.0041126191 | −0.80 | 1.99 × 10−2 | −0.92 | 4.84 × 10−3 | LTBP4 |
| chr20.0031210733 | 1.20 | 2.29 × 10−2 | 1.36 | 2.68 × 10−2 | |
| chr20.0052825772 | −1.35 | 1.91 × 10−3 | −1.31 | 3.11 × 10−3 | PFDN4 |
| chr20.0055369320 | −1.24 | 9.46 × 10−4 | −1.68 | 1.37 × 10−4 | |
| chr20.0060501154 | 2.02 | 6.08 × 10−4 | 1.87 | 9.34 × 10−4 | CDH4 |
| chr21.0030689317 | −0.78 | 4.49 × 10−3 | −0.86 | 1.16 × 10−3 | BACH1 |
| chr22.0050332646 | −1.23 | 5.83 × 10−3 | −1.35 | 1.32 × 10−3 |
| Chromosome | MB | MT | Muscle | Blood | ||||
|---|---|---|---|---|---|---|---|---|
| Significant CpGs | Enrichment p-Value | Significant CpGs | Enrichment p-Value | Significant CpGs | Enrichment p-Value | Significant CpGs | Enrichment p-Value | |
| 1 | 0 | 1.000 | 1 | 1.000 | 1 | 1.000 | 10 | 1.000 |
| 2 | 26 | 0.997 | 103 | 1.000 | 84 | 0.361 | 312 | 1.000 |
| 3 | 45 | 4.81 ×10−3 | 112 | 0.146 | 77 | 9.94 × 10−3 | 650 | 2.20 × 10−16 |
| 4 | 3 | 1.000 | 9 | 1.000 | 7 | 1.000 | 21 | 1.000 |
| 5 | 14 | 0.998 | 82 | 0.863 | 64 | 0.082 | 222 | 1.000 |
| 6 | 29 | 0.348 | 98 | 0.209 | 109 | 1.22 × 10−12 | 544 | 2.20 × 10−16 |
| 7 | 8 | 1.000 | 23 | 1.000 | 23 | 1.000 | 136 | 1.000 |
| 8 | 9 | 1.000 | 26 | 1.000 | 28 | 0.999 | 169 | 1.000 |
| 9 | 18 | 0.921 | 54 | 1.000 | 22 | 1.000 | 208 | 1.000 |
| 10 | 13 | 0.999 | 35 | 1.000 | 4 | 1.000 | 76 | 1.000 |
| 11 | 33 | 0.103 | 101 | 0.092 | 58 | 0.196 | 618 | 2.20 × 10−16 |
| 12 | 76 | 2.20 × 10−16 | 204 | 2.20 × 10−16 | 67 | 7.43 × 10−3 | 519 | 2.20 × 10−16 |
| 13 | 21 | 3.73 × 10−2 | 104 | 7.37 × 10−14 | 61 | 9.60 × 10−9 | 344 | 2.20 × 10−16 |
| 14 | 41 | 3.31 × 10−7 | 137 | 2.20 × 10−16 | 68 | 6.01 × 10−8 | 448 | 2.20 × 10−16 |
| 15 | 31 | 1.63 × 10−3 | 130 | 2.20 × 10−16 | 72 | 6.96 × 10−9 | 298 | 3.44 × 10−9 |
| 16 | 20 | 0.835 | 93 | 0.111 | 68 | 3.04 × 10−3 | 369 | 1.37 × 10−5 |
| 17 | 26 | 0.564 | 86 | 0.659 | 56 | 0.329 | 300 | 0.912 |
| 18 | 35 | 4.65 × 10−8 | 107 | 2.20 × 10−16 | 47 | 1.42 × 10−5 | 341 | 2.20 × 10−16 |
| 19 | 9 | 1.000 | 47 | 1.000 | 25 | 1.000 | 170 | 1.000 |
| 20 | 47 | 4.59 × 10−11 | 153 | 2.20 × 10−16 | 64 | 4.93 × 10−8 | 484 | 2.20 × 10−16 |
| 21 | 2 | 0.995 | 6 | 1.000 | 2 | 1.000 | 9 | 1.000 |
| 22 | 19 | 0.101 | 63 | 1.09 × 10−2 | 31 | 0.257 | 293 | 2.20 × 10−16 |
| Total | 525 | 1774 | 1038 | 6541 | ||||
| TSS | LogFC | FDR Corrected p-Value | Gene | Class |
|---|---|---|---|---|
| chr16:51277965 | –0.85 | 3.82 × 10−4 | AC137527.2 | Pseudogene |
| chr13:115039303 | 0.20 | 1.97 × 10−3 | MIR4502 | miRNA |
| chr17:34397734 | 0.39 | 4.41 × 10−2 | CCL18 | Protein coding |
| TSS | LogFC | FDR Corrected p-Value | Gene | Class |
|---|---|---|---|---|
| chr17:73070401 | 0.59 | 9.50 × 10−6 | AC111186.1 | Pseudogene |
| chr17:75148756 | 0.36 | 4.01 × 10−4 | RNU4–47P | snRNA |
| chr19:48673949 | 0.60 | 8.84 × 10−4 | ZSWIM9 | Protein coding |
| chr11:46134769 | 0.55 | 1.51 × 10−3 | AC024475.1 | miRNA |
| chr4:111866955 | 0.30 | 1.81 × 10−3 | LYPLA1P2 | Pseudogene |
| chr12:7072409 | 0.25 | 6.45 × 10−3 | U47924.27 | lincRNA |
| chr1:242187356 | –0.14 | 6.93 × 10−3 | RNU6–1139P | snRNA |
| chr12:7072408 | 0.25 | 7.37 × 10−3 | EMG1 | Protein coding |
| chr11:93971316 | 1.04 | 1.67 × 10−2 | RP11–680H20.2 | lincRNA |
| chr2:47454056 | –0.67 | 4.96 × 10−2 | AC106869.2 | lincRNA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robinson, K.G.; Marsh, A.G.; Lee, S.K.; Hicks, J.; Romero, B.; Batish, M.; Crowgey, E.L.; Shrader, M.W.; Akins, R.E. DNA Methylation Analysis Reveals Distinct Patterns in Satellite Cell–Derived Myogenic Progenitor Cells of Subjects with Spastic Cerebral Palsy. J. Pers. Med. 2022, 12, 1978. https://doi.org/10.3390/jpm12121978
Robinson KG, Marsh AG, Lee SK, Hicks J, Romero B, Batish M, Crowgey EL, Shrader MW, Akins RE. DNA Methylation Analysis Reveals Distinct Patterns in Satellite Cell–Derived Myogenic Progenitor Cells of Subjects with Spastic Cerebral Palsy. Journal of Personalized Medicine. 2022; 12(12):1978. https://doi.org/10.3390/jpm12121978
Chicago/Turabian StyleRobinson, Karyn G., Adam G. Marsh, Stephanie K. Lee, Jonathan Hicks, Brigette Romero, Mona Batish, Erin L. Crowgey, M. Wade Shrader, and Robert E. Akins. 2022. "DNA Methylation Analysis Reveals Distinct Patterns in Satellite Cell–Derived Myogenic Progenitor Cells of Subjects with Spastic Cerebral Palsy" Journal of Personalized Medicine 12, no. 12: 1978. https://doi.org/10.3390/jpm12121978