
Cerebral Oxygen Delivery and Consumption in Brain-Injured Patients
 , , and
, , and
Abstract
:1. Introduction
2. Oxygen Delivery and Autoregulation
3. Oxygen Consumption
4. Oxygen in the Cells
5. Hyperventilation/Hypoventilation
6. A Brief Search for “4-H” Factors Affecting CRMO2 and Cellular Oxygen Balance
6.1. Hypoxia
- Reduced brain tissue oxygenation is a predictor of poor outcome following severe traumatic brain injury.
- Hypoxic–ischemic brain injury (HIBI) is associated with significant mortality and morbidity [91].
- The LOCO2 study documented that targeting lower PaO2 improves outcomes in patients with acute respiratory distress syndrome (ARDS) [92].
- Secondary hypoxia is connected with extended production of cytokines in CSF and superior elevation of serum biomarkers such as myelin-basic protein (MBP) and S100 [94].
- The MBP, S100 and neuron-specific enolase (NSE) biomarkers are more elevated in patients with hypoxia and unfavorable outcomes (Extended Glasgow Outcome Coma Score (GOSE) 1–4) [94]
- HIBI, as a two-hit model, is an effect of primary and secondary ischemic/hypoxic damage predisposing to overall devastating severe injury of neurovascular units [91]
- Secondary brain hypoxia is connected with de novo neuronal and astroglial injury. Importantly, secondary hypoxia is associated with cerebral proinflammatory response but not parallel cerebral endothelial injury [91].
- Protocols based on PbtO2 and ICP monitoring significantly decrease cerebral hypoxia time after TBI [95].
- Acute intermittent hypoxia (AIH) and task-specific training (TST) may synergistically improve motor functions after central nervous system injury [96].
6.2. Hyperoxia
- Hyperoxia is associated with higher mortality and worse short-term functional outcomes, especially in patients who receive uncontrolled oxygen delivery during the first 24 h after brain injury (probably because of hyperoxia-induced oxygen-free radical toxicity with or without vasoconstriction) [123].
- Potential toxicity of a high oxygen concentration (patients receiving FiO2 of more than 0.6).
- Previous studies documented that higher inspired oxygen concentration is associated with acute lung injury, with mild to severe diffuse alveolar damage (DAD) [124].
- High oxygen levels within 72 h after aneurysmal rupture is an uninfluenced predictor of cerebral vasospasm [125].
- In addition, liberal oxygen therapy increased 30-day mortality compared with conservative therapy [126].
- Controversial high-dose oxygen therapy recommendations to reduce surgical site infections (SSIs) by World Health Organization global guidelines for the prevention of surgical site infection [127].
- Hyperoxemia may reduce cardiac output and increase systemic vascular resistance in patients with cardiovascular failure [128].
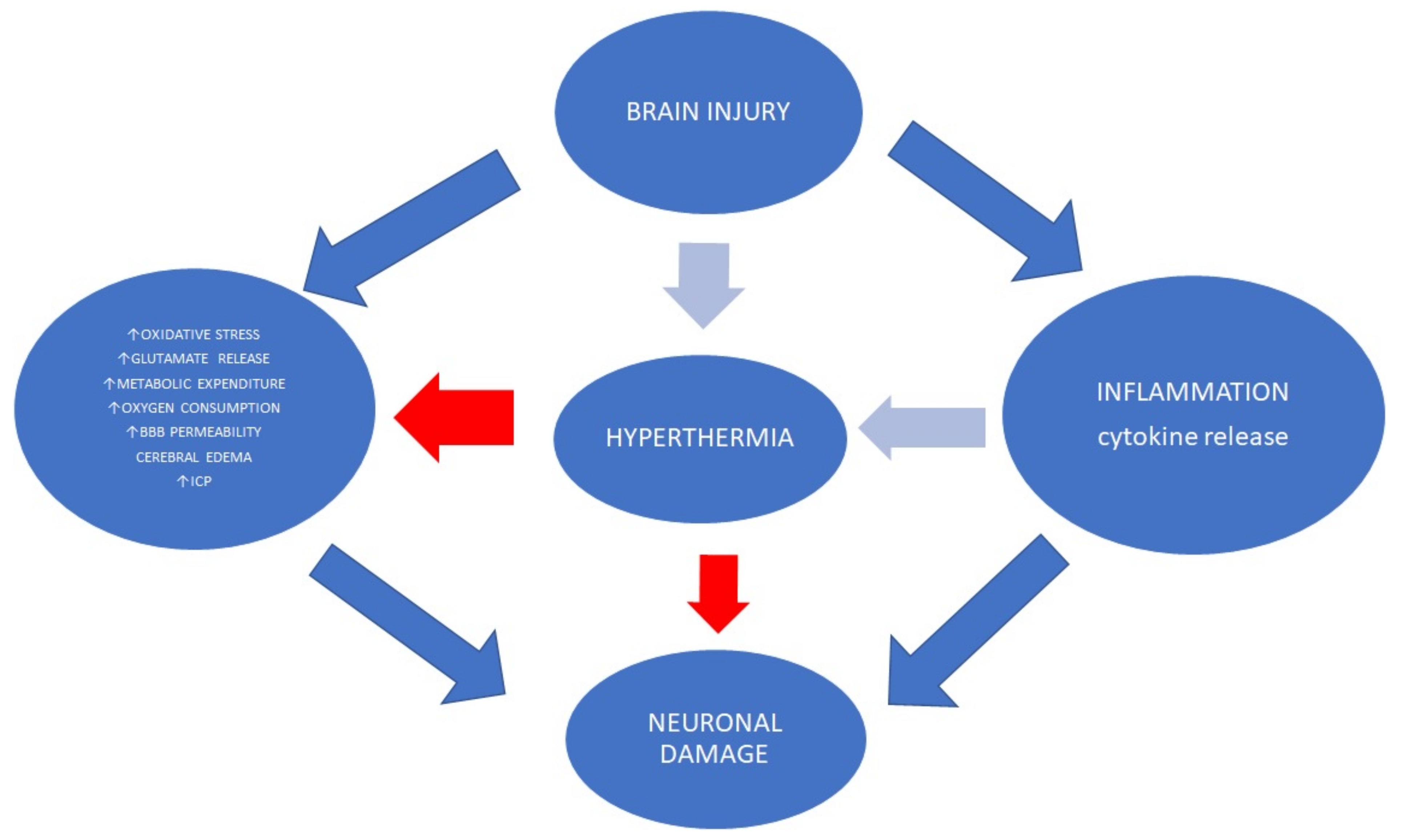
6.3. Hyperthermia
- Up to 50% of patients after acute brain injury experience fever during hospitalization [147].
- Brain temperature variations (>1 °C) are associated with poor functional outcomes [148].
- In sum, higher body temperature is associated with elevated metabolic demand and endogenous stress levels, blood pressure level changes, increases in cardiac output and heart rate, hyperventilation, the synaptic release of excitatory amino acids, increased ICP levels, ischemic cortical depolarizations, and BBB breakdown [146,149,150,151,152,153,154].
- Hyperthermia without oxygen delivery mismatch does not seem to induce significant neurochemical alterations such as glucose, lactate, pyruvate and glutamate levels [151].
- PbtO2 may be an important element to be monitored during a high body temperature episode to provide a view into oxygen metabolism in the brain [155].
- PbtO2 variations are observed under increased temperature increases in severe TBI patients. PbtO2 may rise on average in every third and decrease in every sixth episode of high temperature. Recent data have documented that the PbtO2 slope may occur simultaneously with CPP and MAP reduction [156].
6.4. Hypothermia
- Therapeutic hypothermia is a crucial component of current clinical practice guidelines.
- Therapeutic hypothermia uses different cooling methods to maintain brain temperature at target levels.
- Recently published data do not promote early prophylactic hypothermia within the first 6 h after damage in TBI patients [177].
- Recent meta-analyses have documented the importance of temperature measurement to avoid hypothermia in prehospital management [176].
7. Future Therapies
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Semenza, G.L. Life with Oxygen. Science 2007, 318, 62–64. [Google Scholar] [CrossRef] [PubMed]
- Özugur, S.; Kunz, L.; Straka, H. Relationship between oxygen consumption and neuronal activity in a defined neural circuit. BMC Biol. 2020, 18, 76. [Google Scholar] [CrossRef] [PubMed]
- Roy, C.S.; Sherrington, C.S. On the Regulation of the Blood-supply of the Brain. J. Physiol. 1890, 11, 85–158. [Google Scholar] [CrossRef]
- Maas, A.I.R.; Menon, D.K.; Adelson, P.D.; Andelic, N.; Bell, M.J.; Belli, A.; Bragge, P.; Brazinova, A.; Büki, A.; Chesnut, R.M.; et al. Traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017, 16, 987–1048. [Google Scholar] [CrossRef] [Green Version]
- Chesnut, R.M.; Marshall, L.F.; Klauber, M.R.; Blunt, B.A.; Baldwin, N.; Eisenberg, H.M.; Jane, J.A.; Marmarou, A.; Foulkes, M.A. The Role of Secondary Brain Injury in Determining Outcome from Severe Head Injury. J. Trauma Inj. Infect. Crit. Care 1993, 34, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Volpi, P.C.; Robba, C.; Rota, M.; Vargiolu, A.; Citerio, G. Trajectories of early secondary insults correlate to outcomes of traumatic brain injury: Results from a large, single centre, observational study. BMC Emerg. Med. 2018, 18, 52. [Google Scholar] [CrossRef]
- Maloney-Wilensky, E.; Gracias, V.; Itkin, A.; Hoffman, K.; Bloom, S.; Yang, W.; Christian, S.; Leroux, P.D. Brain tissue oxygen and outcome after severe traumatic brain injury: A systematic review. Crit. Care Med. 2009, 37, 2057–2063. [Google Scholar] [CrossRef]
- Jeremitsky, E.; Omert, L.; Dunham, C.M.; Protetch, J.; Rodriguez, A. Harbingers of Poor Outcome the Day after Severe Brain Injury: Hypothermia, Hypoxia, and Hypoperfusion. J. Trauma Inj. Infect. Crit. Care 2003, 54, 312–319. [Google Scholar] [CrossRef]
- Bogossian, E.G.; Diaferia, D.; Djangang, N.N.; Menozzi, M.; Vincent, J.-L.; Talamonti, M.; Dewitte, O.; Peluso, L.; Barrit, S.; Al Barajraji, M.; et al. Brain tissue oxygenation guided therapy and outcome in non-traumatic subarachnoid hemorrhage. Sci. Rep. 2021, 11, 16235. [Google Scholar] [CrossRef]
- Martini, R.P.; Deem, S.; Treggiari, M.M. Targeting Brain Tissue Oxygenation in Traumatic Brain Injury. Respir. Care 2012, 58, 162–172. [Google Scholar] [CrossRef]
- Chesnut, R.; Aguilera, S.; Buki, A.; Bulger, E.; Citerio, G.; Cooper, D.J.; Arrastia, R.D.; Diringer, M.; Figaji, A.; Gao, G.; et al. A management algorithm for adult patients with both brain oxygen and intracranial pressure monitoring: The Seattle International Severe Traumatic Brain Injury Consensus Conference (SIBICC). Intensiv. Care Med. 2020, 46, 919–929. [Google Scholar] [CrossRef] [Green Version]
- Rakhit, S.; Nordness, M.F.; Lombardo, S.R.; Cook, M.; Smith, L.; Patel, M.B. Management and Challenges of Severe Traumatic Brain Injury. Semin. Respir. Crit. Care Med. 2021, 42, 127–144. [Google Scholar] [CrossRef]
- Sekhon, M.S.; Gooderham, P.; Menon, D.K.; Brasher, P.M.A.; Foster, D.; Cardim, D.; Czosnyka, M.; Smielewski, P.; Gupta, A.K.; Ainslie, P.N.; et al. The Burden of Brain Hypoxia and Optimal Mean Arterial Pressure in Patients With Hypoxic Ischemic Brain Injury After Cardiac Arrest. Crit. Care Med. 2019, 47, 960–969. [Google Scholar] [CrossRef]
- Hyder, F.; Rothman, D.L.; Bennett, M.R. Cortical energy demands of signaling and nonsignaling components in brain are conserved across mammalian species and activity levels. Proc. Natl. Acad. Sci. USA 2013, 110, 3549–3554. [Google Scholar] [CrossRef] [Green Version]
- Rink, C.; Khanna, S. Significance of Brain Tissue Oxygenation and the Arachidonic Acid Cascade in Stroke. Antioxid. Redox Signal 2011, 14, 1889–1903. [Google Scholar] [CrossRef] [Green Version]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic Energy Use and Supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef] [Green Version]
- Gjedde, A. The pathways of oxygen in brain. I. Delivery and metabolism of oxygen. In Advances in Experimental Medicine and Biology; Springer: Boston, MA, USA, 2005; p. 566. [Google Scholar]
- Bain, A.R.; Morrison, S.; Ainslie, P.N. Cerebral oxygenation and hyperthermia. Front. Physiol. 2014, 5, 92. [Google Scholar] [CrossRef] [Green Version]
- Paulson, O.B.; Strandgaard, S.; Edvinsson, L. Cerebral autoregulation. Cerebrovasc. Brain Metab. Rev. 1990, 2, 161–192. [Google Scholar]
- Cipolla, M.J.; Osol, G. Vascular Smooth Muscle Actin Cytoskeleton in Cerebral Artery Forced Dilatation. Stroke 1998, 29, 1223–1228. [Google Scholar] [CrossRef] [Green Version]
- Baron, J.-C. Perfusion Thresholds in Human Cerebral Ischemia: Historical Perspective and Therapeutic Implications. Cerebrovasc. Dis. 2001, 11 (Suppl. 1), 2–8. [Google Scholar] [CrossRef]
- Tas, J.; Beqiri, E.; van Kaam, R.C.; Czosnyka, M.; Donnelly, J.; Haeren, R.H.; van der Horst, I.C.; Hutchinson, P.J.; van Kuijk, S.M.; Liberti, A.L.; et al. Targeting Autoregulation-Guided Cerebral Perfusion Pressure after Traumatic Brain Injury (COGiTATE): A Feasibility Randomized Controlled Clinical Trial. J. Neurotrauma 2021, 38, 2790–2800. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Wu, H.-B.; Yan, Y.-F.; Liu, M.; Wang, E.-S. Mortality and Outcome Comparison Between Brain Tissue Oxygen Combined with Intracranial Pressure/Cerebral Perfusion Pressure–Guided Therapy and Intracranial Pressure/Cerebral Perfusion Pressure–Guided Therapy in Traumatic Brain Injury: A Meta-Analysis. World Neurosurg. 2017, 100, 118–127. [Google Scholar] [CrossRef] [PubMed]
- McDowall, D.G. Interrelationships between blood oxygen tensions and cerebral blood flow. Oxyg. Meas. Blood Tissues 1966, 4, 205–219. [Google Scholar]
- Attwell, D.; Buchan, A.M.; Charpak, S.; Lauritzen, M.J.; MacVicar, B.A.; Newman, E.A. Glial and neuronal control of brain blood flow. Nature 2010, 468, 232–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muoio, V.; Persson, P.B.; Sendeski, M.M. The neurovascular unit—concept review. Acta Physiol. 2014, 210, 790–798. [Google Scholar] [CrossRef]
- Sokoloff, L. Energetics of Functional Activation in Neural Tissues. Neurochem. Res. 1999, 24, 321–329. [Google Scholar] [CrossRef]
- Hattori, N.; Huang, S.-C.; Wu, H.-M.; Yeh, E.; Glenn, T.C.; Vespa, P.M.; McArthur, D.; E Phelps, M.; A Hovda, D.; Bergsneider, M. Correlation of regional metabolic rates of glucose with glasgow coma scale after traumatic brain injury. J. Nucl. Med. 2003, 44, 1709. [Google Scholar]
- Bergsneider, M.; Hovda, D.A.; Lee, S.M.; Kelly, D.F.; McArthur, D.; Vespa, P.M.; Lee, J.H.; Huang, S.-C.; Martin, N.; Phelps, M.E.; et al. Dissociation of Cerebral Glucose Metabolism and Level of Consciousness During the Period of Metabolic Depression Following Human Traumatic Brain Injury. J. Neurotrauma 2000, 17, 389–401. [Google Scholar] [CrossRef]
- Bergsneider, M.; Hovda, D.A.; Shalmon, E.; Kelly, D.F.; Vespa, P.M.; Martin, N.A.; Phelps, M.E.; McArthur, D.L.; Caron, M.J.; Kraus, J.F.; et al. Cerebral hyperglycolysis following severe traumatic brain injury in humans: A positron emission tomography study. J. Neurosurg. 1997, 86, 241–251. [Google Scholar] [CrossRef]
- Obrist, W.D.; Langfitt, T.W.; Jaggi, J.L.; Cruz, J.; Gennarelli, T.A. Cerebral blood flow and metabolism in comatose patients with acute head injury: Relationship to intracranial hypertension. J. Neurosurg. 1984, 61, 241–253. [Google Scholar] [CrossRef]
- Soustiel, J.F.; Glenn, T.C.; Shik, V.; Boscardin, J.; Mahamid, E.; Zaaroor, M. Monitoring of Cerebral Blood Flow and Metabolism in Traumatic Brain Injury. J. Neurotrauma 2005, 22, 955–965. [Google Scholar] [CrossRef]
- Cold, G.E. Cerebral Metabolic Rate of Oxygen (CMRO2) in the Acute Phase of Brain Injury. Acta Anaesthesiol. Scand. 1978, 22, 249–256. [Google Scholar] [CrossRef]
- Shulman, R.G.; Hyder, F.; Rothman, D.L. Lactate efflux and the neuroenergetic basis of brain function. NMR Biomed. 2001, 14, 389–396. [Google Scholar] [CrossRef]
- Thompson Jeffrey, K.; Peterson Matthew, R.; Freeman Ralph, D. Single-Neuron Activity and Tissue Oxygenation in the Cerebral Cortex. Science 2003, 299, 1070–1072. [Google Scholar] [CrossRef] [Green Version]
- Hyder, F.; Kida, I.; Behar, K.L.; Kennan, R.P.; Maciejewski, P.K.; Rothman, D.L. Quantitative functional imaging of the brain: Towards mapping neuronal activity by BOLD fMRI. NMR Biomed. 2001, 14, 413–431. [Google Scholar] [CrossRef]
- Hochachka, P.W.; Buck, L.T.; Doll, C.J.; Land, S.C. Unifying theory of hypoxia tolerance: Molecular/metabolic defense and rescue mechanisms for surviving oxygen lack. Proc. Natl. Acad. Sci. USA 1996, 93, 9493–9498. [Google Scholar] [CrossRef] [Green Version]
- Boyer, P.D. The Atp Synthase—A Splendid Molecular Machine. Annu. Rev. Biochem. 1997, 66, 717–749. [Google Scholar] [CrossRef] [Green Version]
- Wu, I.C.; Ohsawa, I.; Fuku, N.; Tanaka, M. Metabolic analysis of 13C-labeled pyruvate for noninvasive assessment of mitochondrial function. Ann. N. Y. Acad. Sci. 2010, 1201, 111–120. [Google Scholar] [CrossRef]
- Wellen, K.E.; Thompson, C.B. Cellular metabolic stress: Considering how cells respond to nutrient excess. Mol. Cell 2010, 40, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Sas, K.; Robotka, H.; Toldi, J.; Vécsei, L. Mitochondria, metabolic disturbances, oxidative stress and the kynurenine system, with focus on neurodegenerative disorders. J. Neurol. Sci. 2007, 257, 221–239. [Google Scholar] [CrossRef]
- Diringer, M.N.; Yundt, K.; Videen, T.O.; Adams, R.E.; Zazulia, A.R.; Deibert, E.; Aiyagari, V.; Dacey, R.G.; Grubb, R.L.; Powers, W.J. No reduction in cerebral metabolism as a result of early moderate hyperventilation following severe traumatic brain injury. J. Neurosurg. 2000, 92, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Guo, Q.; Wang, E. Hyperventilation in neurological patients: From physiology to outcome evidence. Curr. Opin. Anesthesiol. 2019, 32, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Mas, A.; Saura, P.; Joseph, D.; Blanch, L.; Baigorri, F.; Artigas, A.; Fernandez, R. Effect of acute moderate changes in PaCO2 on global hemodynamics and gastric perfusion. Crit. Care Med. 2000, 28, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Howarth, C.; Sutherland, B.A.; Choi, H.B.; Martin, C.; Lind, B.L.; Khennouf, L.; LeDue, J.M.; Pakan, J.M.; Ko, R.W.; Ellis-Davies, G.; et al. A Critical Role for Astrocytes in Hypercapnic Vasodilation in Brain. J. Neurosci. 2017, 37, 2403–2414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carney, N.; Totten, A.M.; O’Reilly, C.; Ullman, J.S.; Hawryluk, G.W.; Bell, M.J.; Bratton, S.L.; Chesnut, R.; Harris, O.A.; Kissoon, N.; et al. Guidelines for the Management of Severe Traumatic Brain Injury, Fourth Edition. Neurosurgery 2017, 80, 6–15. [Google Scholar] [CrossRef]
- Oddo, M.; Bösel, J. Monitoring of brain and systemic oxygenation in neurocritical care patients. Neurocritical Care 2014, 21, 103–120. [Google Scholar] [CrossRef]
- Chen, R.; Lai, U.H.; Zhu, L.; Singh, A.; Ahmed, M.; Forsyth, N.R. Reactive Oxygen Species Formation in the Brain at Different Oxygen Levels: The Role of Hypoxia Inducible Factors. Front. Cell Dev. Biol. 2018, 6, 132. [Google Scholar] [CrossRef] [Green Version]
- Banasiak, K.J.; Xia, Y.; Haddad, G.G. Mechanisms underlying hypoxia-induced neuronal apoptosis. Prog. Neurobiol. 2000, 62, 215–249. [Google Scholar] [CrossRef]
- Chang, E.; Hornick, K.; Fritz, K.I.; Mishra, O.P.; Delivoria-Papadopoulos, M. Effect of Hyperoxia on Cortical Neuronal Nuclear Function and Programmed Cell Death Mechanisms. Neurochem. Res. 2007, 32, 1142–1149. [Google Scholar] [CrossRef]
- Serocki, M.; Bartoszewska, S.; Janaszak-Jasiecka, A.; Ochocka, R.J.; Collawn, J.F.; Bartoszewski, R. miRNAs regulate the HIF switch during hypoxia: A novel therapeutic target. Angiogenesis 2018, 21, 183–202. [Google Scholar] [CrossRef] [Green Version]
- Koh, M.Y.; Lemos, R.; Liu, X.; Powis, G. The hypoxia-associated factor switches cells from HIF-1α-to HIF-2α-dependent signaling promoting stem cell characteristics, aggressive tumor growth and invasion. Cancer Res. 2011, 71, 4015–4027. [Google Scholar] [CrossRef] [Green Version]
- Masamoto, K.; Tanishita, K. Oxygen Transport in Brain Tissue. J. Biomech. Eng. 2009, 131, 074002. [Google Scholar] [CrossRef]
- Taguchi, H.; Heistad, D.D.; Kitazono, T.; Faraci, F.M. ATP-sensitive K+ channels mediate dilatation of cerebral arterioles during hypoxia. Circ. Res. 1994, 74, 1005–1008. [Google Scholar] [CrossRef]
- Johnston, A.J.; Steiner, L.A.; Gupta, A.K.; Menon, D.K. Cerebral oxygen vasoreactivity and cerebral tissue oxygen reactivity. Br. J. Anaesth. 2003, 90, 774–786. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; LaManna, J.C. Chronic hypoxia and the cerebral circulation. J. Appl. Physiol. 2006, 100, 725–730. [Google Scholar] [CrossRef]
- Boero, J.A.; Ascher, J.; Arregui, A.; Rovainen, C.; Woolsey, T.A. Increased brain capillaries in chronic hypoxia. J. Appl. Physiol. 1999, 86, 1211–1219. [Google Scholar] [CrossRef]
- Torres-Cuevas, I.; Parra-Llorca, A.; Sánchez-Illana, Á.; Nuñez-Ramiro, A.; Kuligowski, J.; Cháfer-Pericás, C.; Cernada, M.; Escobar, J.; Vento, M. Oxygen and oxidative stress in the perinatal period. Redox Biol. 2017, 12, 674–681. [Google Scholar] [CrossRef]
- Rocha-Ferreira, E.; Hristova, M. Plasticity in the Neonatal Brain following Hypoxic-Ischaemic Injury. Neural Plast. 2016, 2016, 4901014. [Google Scholar] [CrossRef] [Green Version]
- Sekhon, M.S.; Ainslie, P.N.; Griesdale, D.E. Clinical pathophysiology of hypoxic ischemic brain injury after cardiac arrest: A “two-hit” model. Crit. Care 2017, 21, 90. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Yoshioka, H.; Kim, G.S.; Jung, J.E.; Okami, N.; Sakata, H.; Maier, C.M.; Narasimhan, P.; Goeders, C.E.; Chan, P.H. Oxidative Stress in Ischemic Brain Damage: Mechanisms of Cell Death and Potential Molecular Targets for Neuroprotection. Antioxid. Redox Signal. 2011, 14, 1505–1517. [Google Scholar] [CrossRef] [Green Version]
- Coimbra-Costa, D.; Alva, N.; Duran, M.; Carbonell, T.; Rama, R. Oxidative stress and apoptosis after acute respiratory hypoxia and reoxygenation in rat brain. Redox Biol. 2017, 12, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Peña, F.; Ramirez, J.-M. Hypoxia-induced changes in neuronal network properties. Mol. Neurobiol. 2005, 32, 251–283. [Google Scholar] [CrossRef]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Dengler, V.L.; Galbraith, M.D.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chávez, J.C.; Agani, F.; Pichiule, P.; LaManna, J.C. Expression of hypoxia-inducible factor-1α in the brain of rats during chronic hypoxia. J. Appl. Physiol. 2000, 89, 1937–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukyanova, L.D.; Kirova, Y.I. Mitochondria-controlled signaling mechanisms of brain protection in hypoxia. Front. Neurosci. 2015, 9, 320. [Google Scholar] [CrossRef] [Green Version]
- Brix, B.; Mesters, J.R.; Pellerin, L.; Jöhren, O. Endothelial cell-derived nitric oxide enhances aerobic glycolysis in astrocytes via HIF-1α-mediated target gene activation. J. Neurosci. 2012, 32, 9727–9735. [Google Scholar] [CrossRef]
- Koh, M.Y.; Powis, G. Passing the baton: The HIF switch. Trends Biochem. Sci. 2012, 37, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Zhong, J.; Chang, R.; Hu, H.; Pandey, A.; Semenza, G.L. Hsp70 and CHIP Selectively Mediate Ubiquitination and Degradation of Hypoxia-inducible Factor (HIF)-1α but Not HIF-2α 2. J. Biol. Chem. 2010, 285, 3651–3663. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.V.; Semenza, G.L. RACK1 vs. HSP90: Competition for HIF-1α degradation vs. stabilization. Cell Cycle 2007, 6, 656–659. [Google Scholar] [CrossRef]
- Liu, Y.V.; Baek, J.H.; Zhang, H.; Diez, R.; Cole, R.N.; Semenza, G.L. RACK1 competes with HSP90 for binding to HIF-1α and is required for O2-independent and HSP90 inhibitor-induced degradation of HIF-1α. Mol. Cell 2007, 25, 207–217. [Google Scholar] [CrossRef] [Green Version]
- Ravi, R.; Mookerjee, B.; Bhujwalla, Z.M.; Sutter, C.H.; Artemov, D.; Zeng, Q.; Dillehay, L.E.; Madan, A.; Semenza, G.L.; Bedi, A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1α. Genes Dev. 2000, 14, 34–44. [Google Scholar] [CrossRef]
- Bhattacharya, R.; SenBanerjee, S.; Lin, Z.; Mir, S.; Hamik, A.; Wang, P.; Mukherjee, P.; Mukhopadhyay, D.; Jain, M.K. Inhibition of Vascular Permeability Factor/Vascular Endothelial Growth Factor-mediated Angiogenesis by the Kruppel-like Factor KLF2. J. Biol. Chem. 2005, 280, 28848–28851. [Google Scholar] [CrossRef]
- Kawanami, D.; Mahabeleshwar, G.H.; Lin, Z.; Atkins, G.B.; Hamik, A.; Haldar, S.M.; Maemura, K.; LaManna, J.C.; Jain, M.K. Kruppel-like Factor 2 Inhibits Hypoxia-inducible Factor 1α Expression and Function in the Endothelium. J. Biol. Chem. 2009, 284, 20522–20530. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef]
- Davis, G.M.; Haas, M.A.; Pocock, R. MicroRNAs: Not “fine-tuners” but key regulators of neuronal development and function. Front. Neurol. 2015, 6, 245. [Google Scholar] [CrossRef] [Green Version]
- Xiao, S.; Ma, Y.; Zhu, H.; Sun, H.; Yin, Y.; Feng, G. miRNA functional synergistic network analysis of mice with ischemic stroke. Neurol. Sci. 2015, 36, 143–148. [Google Scholar] [CrossRef]
- Chan, S.Y.; Loscalzo, J. MicroRNA-210: A unique and pleiotropic hypoxamir. Cell Cycle 2010, 9, 1072–1083. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.-Y.; Zeng, L.; Liu, J.; Wang, Y.; Wang, L.; Weng, S.; Tang, Y.; Zheng, C.; Cheng, Q.; Chen, S. MicroRNA-210 as a novel blood biomarker in acute cerebral ischemia. Front. Biosci. 2011, E3, 1265–1272. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Zhou, X.-Y.; Zhou, X.-G.; Cheng, R.; Liu, H.-Y.; Li, Y. Neuroprotective effects of microRNA-210 on hypoxic-ischemic encephalopathy. BioMed Res. Int. 2013, 2013, 350419. [Google Scholar] [CrossRef] [Green Version]
- Tan, K.S.; Armugam, A.; Sepramaniam, S.; Lim, K.Y.; Setyowati, K.D.; Wang, C.W.; Jeyaseelan, K. Expression Profile of MicroRNAs in Young Stroke Patients. PLoS ONE 2009, 4, e7689. [Google Scholar] [CrossRef] [Green Version]
- Minhas, G.; Mathur, D.; Ragavendrasamy, B.; Sharma, N.K.; Paanu, V.; Anand, A. Hypoxia in CNS Pathologies: Emerging Role of miRNA-Based Neurotherapeutics and Yoga Based Alternative Therapies. Front. Neurosci. 2017, 11, 386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobryshev, P.; Bagaeva, T.; Filaretova, L. Ischemic preconditioning attenuates gastric ischemia-reperfusion injury through involvement of glucocorticoids. J. Physiol. Pharmacol. 2009, 60 (Suppl. 7), 155–160. [Google Scholar]
- Rybnikova, E.A.; Mironova, V.I.; Pivina, S.G.; Ordyan, N.E.; Tulkova, E.I.; Samoilov, M.O. Hormonal Mechanisms of Neuroprotective Effects of the Mild Hypoxic Preconditioning in Rats, 1st ed.; Springer Nature BV: Berlin/Heidelberg, Germany, 2008; p. 239. [Google Scholar]
- Davis, C.; Hackett, P. Advances in the prevention and treatment of high altitude illness. Emerg. Med. Clin. 2017, 35, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Rybnikova, E.; Mironova, V.; Pivina, S.; Tulkova, E.; Ordyan, N.; Nalivaeva, N.; Turner, A.; Samoilov, M. Involvement of the hypothalamic-pituitary-adrenal axis in the antidepressant-like effects of mild hypoxic preconditioning in rats. Psychoneuroendocrinology 2007, 32, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Vanderhaeghen, T.; Beyaert, R.; Libert, C. Bidirectional Crosstalk Between Hypoxia Inducible Factors and Glucocorticoid Signalling in Health and Disease. Front. Immunol. 2021, 12, 684085. [Google Scholar] [CrossRef]
- Kodama, T.; Shimizu, N.; Yoshikawa, N.; Makino, Y.; Ouchida, R.; Okamoto, K.; Hisada, T.; Nakamura, H.; Morimoto, C.; Tanaka, H. Role of the Glucocorticoid Receptor for Regulation of Hypoxia-dependent Gene Expression. J. Biol. Chem. 2003, 278, 33384–33391. [Google Scholar] [CrossRef] [Green Version]
- Lim, W.; Park, C.; Shim, M.K.; Lee, Y.H.; Lee, Y.M.; Lee, Y. Glucocorticoids suppress hypoxia-induced COX-2 and hypoxia inducible factor-1α expression through the induction of glucocorticoid-induced leucine zipper. Br. J. Pharmacol. 2014, 171, 735–745. [Google Scholar] [CrossRef] [Green Version]
- Hoiland, R.L.; Ainslie, P.N.; Wellington, C.L.; Cooper, J.; Stukas, S.; Thiara, S.; Foster, D.; Fergusson, N.A.; Conway, E.M.; Menon, D.K.; et al. Brain Hypoxia Is Associated With Neuroglial Injury in Humans Post–Cardiac Arrest. Circ. Res. 2021, 129, 583–597. [Google Scholar] [CrossRef]
- Barrot, L.; Asfar, P.; Mauny, F.; Winiszewski, H.; Montini, F.; Badie, J.; Quenot, J.-P.; Pili-Floury, S.; Bouhemad, B.; Louis, G.; et al. Liberal or Conservative Oxygen Therapy for Acute Respiratory Distress Syndrome. N. Engl. J. Med. 2020, 382, 999–1008. [Google Scholar] [CrossRef]
- Bardt, T.F.; Unterberg, A.W.; Härtl, R.; Kiening, K.L.; Schneider, G.H.; Lanksch, W.R. Monitoring of brain tissue PO2 in traumatic brain injury: Effect of cerebral hypoxia on outcome. In Intracranial Pressure and Neuromonitoring in Brain Injury; Marmarou, A., Bullock, R., Avezaat, C., Baethmann, A., Becker, D., Brock, M., Hoff, J., Nagai, H., Reulen, H.-J., Teasdale, G., Eds.; Springer: Vienna, Austria, 1998; pp. 153–156. [Google Scholar]
- Yan, E.B.; Satgunaseelan, L.; Paul, E.; Bye, N.; Nguyen, P.; Agyapomaa, D.; Kossmann, T.; Rosenfeld, J.V.; Morganti-Kossmann, C. Post-Traumatic Hypoxia Is Associated with Prolonged Cerebral Cytokine Production, Higher Serum Biomarker Levels, and Poor Outcome in Patients with Severe Traumatic Brain Injury. J. Neurotrauma 2013, 31, 618–629. [Google Scholar] [CrossRef] [Green Version]
- Okonkwo, D.O.; Shutter, L.; Moore, C.; Temkin, N.R.; Puccio, A.M.; Madden, C.J.; Andaluz, N.; Chesnut, R.; Bullock, M.R.; Grant, G.A.; et al. Brain Oxygen Optimization in Severe Traumatic Brain Injury Phase-II. Crit. Care Med. 2017, 45, 1907–1914. [Google Scholar] [CrossRef]
- Welch, J.F.; Sutor, T.W.; Vose, A.K.; Perim, R.R.; Fox, E.J.; Mitchell, G.S. Synergy between Acute Intermittent Hypoxia and Task-Specific Training. Exerc. Sport Sci. Rev. 2020, 48, 125–132. [Google Scholar] [CrossRef]
- Gore, A.; Muralidhar, M.; Espey, M.G.; Degenhardt, K.; Mantell, L.L. Hyperoxia sensing: From molecular mechanisms to significance in disease. J. Immunotoxicol. 2010, 7, 239–254. [Google Scholar] [CrossRef]
- Bin-Jaliah, I.; Haffor, A.-S. Ultrastructural Morphological Alterations during Hyperoxia Exposure in Relation to Glutathione Peroxidase Activity and Free Radicals Productions in the Mitochondria of the Cortical Brain. Int. J. Morphol. 2018, 36, 1310–1315. [Google Scholar] [CrossRef] [Green Version]
- Chong, Z.-Z.; Lin, S.H.; Li, F.; Maiese, K. The Sirtuin Inhibitor Nicotinamide Enhances Neuronal Cell Survival During Acute Anoxic Injury Through AKT, BAD, PARP, and Mitochondrial Associated. Curr. Neurovascular Res. 2005, 2, 271–285. [Google Scholar] [CrossRef]
- Terraneo, L.; Paroni, R.; Bianciardi, P.; Giallongo, T.; Carelli, S.; Gorio, A.; Samaja, M. Brain adaptation to hypoxia and hyperoxia in mice. Redox Biol. 2017, 11, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Felderhoff-Mueser, U.; Bittigau, P.; Sifringer, M.; Jarosz, B.; Korobowicz, E.; Mahler, L.; Piening, T.; Moysich, A.; Grune, T.; Thor, F.; et al. Oxygen causes cell death in the developing brain. Neurobiol. Dis. 2004, 17, 273–282. [Google Scholar] [CrossRef]
- Zhang, Y.; Park, T.S.; Gidday, J.M. Hypoxic preconditioning protects human brain endothelium from ischemic apoptosis by Akt-dependent survivin activation. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2573–H2581. [Google Scholar] [CrossRef] [Green Version]
- Nayak, G.H.; Prentice, H.M.; Milton, S.L. Neuroprotective signaling pathways are modulated by adenosine in the anoxia tolerant turtle. J. Cereb. Blood Flow Metab. 2011, 31, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Mao, M.; Zhiling, W.; Hui, Z.; Shengfu, L.; Dan, Y.; Jiping, H. Cellular levels of TrkB and MAPK in the neuroprotective role of BDNF for embryonic rat cortical neurons against hypoxia in vitro. Int. J. Dev. Neurosci. 2005, 23, 515–521. [Google Scholar]
- Digicaylioglu, M.; Bichet, S.; Marti, H.H.; Wenger, R.H.; Rivas, L.A.; Bauer, C.; Gassmann, M. Localization of specific erythropoietin binding sites in defined areas of the mouse brain. Proc. Natl. Acad. Sci. USA 1995, 92, 3717–3720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, P.E.; Fares, R.P.; Risso, J.-J.; Bonnet, C.; Bouvard, S.; Le-Cavorsin, M.; Georges, B.; Moulin, C.; Belmeguenai, A.; Bodennec, J.; et al. Optimal neuroprotection by erythropoietin requires elevated expression of its receptor in neurons. Proc. Natl. Acad. Sci. USA 2009, 106, 9848–9853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakanaka, M.; Wen, T.-C.; Matsuda, S.; Masuda, S.; Morishita, E.; Nagao, M.; Sasaki, R. In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc. Natl. Acad. Sci. USA 1998, 95, 4635–4640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, S.; Aoyama, M.; Kakita, H.; Hida, H.; Kato, I.; Ito, T.; Goto, T.; Hussein, M.H.; Sawamoto, K.; Togari, H.; et al. Endogenous erythropoietin from astrocyte protects the oligodendrocyte precursor cell against hypoxic and reoxygenation injury. J. Neurosci. Res. 2011, 89, 1566–1574. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C. Nitric oxide and neuronal death. Nitric Oxide 2010, 23, 153–165. [Google Scholar] [CrossRef]
- Garry, P.S.; Ezra, M.; Rowland, M.J.; Westbrook, J.; Pattinson, K.T.S. The role of the nitric oxide pathway in brain injury and its treatment—From bench to bedside. Exp. Neurol. 2015, 263, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.; Wang, S.; Cai, J.; Rao, M.S.; Mattson, M.P. Nitric oxide acts in a positive feedback loop with BDNF to regulate neural progenitor cell proliferation and differentiation in the mammalian brain. Dev. Biol. 2003, 258, 319–333. [Google Scholar] [CrossRef] [Green Version]
- Li, S.-T.; Pan, J.; Hua, X.-M.; Liu, H.; Shen, S.; Liu, J.-F.; Li, B.; Tao, B.-B.; Ge, X.-L.; Wang, X.-H.; et al. Endothelial Nitric Oxide Synthase Protects Neurons against Ischemic Injury through Regulation of Brain-Derived Neurotrophic Factor Expression. CNS Neurosci. Ther. 2014, 20, 154–164. [Google Scholar] [CrossRef]
- Brune, B.; Zhou, J. The role of nitric oxide (NO) in stability regulation of hypoxia inducible factor-1α (HIF-1α). Curr. Med. Chem. 2003, 10, 845–855. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Wang, L.; Asavaritkrai, P.; Noguchi, C.T. Up-regulation of erythropoietin receptor by nitric oxide mediates hypoxia preconditioning. J. Neurosci. Res. 2010, 88, 3180–3188. [Google Scholar] [CrossRef]
- Kasuno, K.; Takabuchi, S.; Fukuda, K.; Kizaka-Kondoh, S.; Yodoi, J.; Adachi, T.; Semenza, G.L.; Hirota, K. Nitric Oxide Induces Hypoxia-inducible Factor 1 Activation That Is Dependent on MAPK and Phosphatidylinositol 3-Kinase Signaling. J. Biol. Chem. 2004, 279, 2550–2558. [Google Scholar] [CrossRef] [Green Version]
- Haase, V.H. The VHL/HIF oxygen-sensing pathway and its relevance to kidney disease. Kidney Int. 2006, 69, 1302–1307. [Google Scholar] [CrossRef] [Green Version]
- Hoehn, T.; Felderhoff-Mueser, U.; Maschewski, K.; Stadelmann, C.; Sifringer, M.; Bittigau, P.; Koehne, P.; Hoppenz, M.; Obladen, M.; Bührer, C. Hyperoxia Causes Inducible Nitric Oxide Synthase-Mediated Cellular Damage to the Immature Rat Brain. Pediatr. Res. 2003, 54, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Zhilyaev, S.Y.; Moskvin, A.N.; Platonova, T.F.; Gutsaeva, D.R.; Churilina, I.V.; Demchenko, I.T. Hyperoxic Vasoconstriction in the Brain Is Mediated by Inactivation of Nitric Oxide by Superoxide Anions. Neurosci. Behav. Physiol. 2003, 33, 783–787. [Google Scholar] [CrossRef]
- Rocha-Ferreira, E.; Rudge, B.; Hughes, M.P.; Rahim, A.A.; Hristova, M.; Robertson, N.J. Immediate Remote Ischemic Postconditioning Reduces Brain Nitrotyrosine Formation in a Piglet Asphyxia Model. Oxidative Med. Cell. Longev. 2016, 2016, 5763743. [Google Scholar] [CrossRef] [Green Version]
- Attaye, I.; Smulders, Y.M.; De Waard, M.C.; Straaten, H.M.O.-V.; Smit, B.; Van Wijhe, M.H.; Musters, R.J.; Koolwijk, P.; Man, A.M.E.S. The effects of hyperoxia on microvascular endothelial cell proliferation and production of vaso-active substances. Intensiv. Care Med. Exp. 2017, 5, 22. [Google Scholar] [CrossRef]
- Demchenko, I.T.; Oury, T.D.; Crapo, J.D.; Piantadosi, C.A. Regulation of the Brain’s Vascular Responses to Oxygen. Circ. Res. 2002, 91, 1031–1037. [Google Scholar] [CrossRef] [Green Version]
- Pham, H.; Vottier, G.; Pansiot, J.; Duong-Quy, S.; Bollen, B.; Dalous, J.; Gallego, J.; Mercier, J.-C.; Dinh-Xuan, A.T.; Bonnin, P.; et al. Inhaled NO prevents hyperoxia-induced white matter damage in neonatal rats. Exp. Neurol. 2014, 252, 114–123. [Google Scholar] [CrossRef]
- Brenner, M.; Stein, D.; Hu, P.; Kufera, J.; Wooford, M.; Scalea, T. Association Between Early Hyperoxia and Worse Outcomes After Traumatic Brain Injury. Arch. Surg. 2012, 147, 1042–1046. [Google Scholar] [CrossRef] [Green Version]
- Davis, W.B.; Rennard, S.I.; Bitterman, P.B.; Crystal, R.G. Pulmonary oxygen toxicity: Early reversible changes in human alveolar structures induced by hyperoxia. N. Engl. J. Med. 1983, 309, 878–883. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, R.A.; Amin, S.N.; Jonathan, S.V.; Tang, A.R.; Lan, M.; Wang, C.; Bastarache, J.A.; Ware, L.B.; Thompson, R.C. Hyperoxemia and Cerebral Vasospasm in Aneurysmal Subarachnoid Hemorrhage. Neurocritical Care 2021, 35, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.K.; Kim, L.H.-Y.; Young, P.J.; Zamiri, N.; Almenawer, S.A.; Jaeschke, R.; Szczeklik, W.; Schünemann, H.J.; Neary, J.D.; Alhazzani, W. Mortality and morbidity in acutely ill adults treated with liberal versus conservative oxygen therapy (IOTA): A systematic review and meta-analysis. Lancet 2018, 391, 1693–1705. [Google Scholar] [CrossRef]
- Suzuki, S. Oxygen administration for postoperative surgical patients: A narrative review. J. Intensive Care 2020, 8, 79. [Google Scholar] [CrossRef] [PubMed]
- Smit, B.; Smulders, Y.M.; van der Wouden, J.C.; Oudemans-van Straaten, H.M.; Spoelstra-de Man, A.M.E. Hemodynamic effects of acute hyperoxia: Systematic review and meta-analysis. Crit. Care 2018, 22, 45. [Google Scholar] [CrossRef] [PubMed]
- Chatzipanteli, K.; Alonso, O.F.; Kraydieh, S.; Dietrich, W.D. Importance of Posttraumatic Hypothermia and Hyperthermia on the Inflammatory Response after Fluid Percussion Brain Injury: Biochemical and Immunocytochemical Studies. J. Cereb. Blood Flow Metab. 2000, 20, 531–542. [Google Scholar] [CrossRef]
- Thompson, H.J.; Tkacs, N.C.; Saatman, K.E.; Raghupathi, R.; McIntosh, T.K. Hyperthermia following traumatic brain injury: A critical evaluation. Neurobiol. Dis. 2003, 12, 163–173. [Google Scholar] [CrossRef]
- Truettner, J.S.; Bramlett, H.M.; Dietrich, W.D. Hyperthermia and Mild Traumatic Brain Injury: Effects on Inflammation and the Cerebral Vasculature. J. Neurotrauma 2017, 35, 940–952. [Google Scholar] [CrossRef]
- Huang, T.; Solano, J.; He, D.; Loutfi, M.; Dietrich, W.D.; Kuluz, J.W. Traumatic Injury Activates MAP Kinases in Astrocytes: Mechanisms of Hypothermia and Hyperthermia. J. Neurotrauma 2009, 26, 1535–1545. [Google Scholar] [CrossRef]
- Kurokawa, H.; Ito, H.; Terasaki, M.; Matsui, H. Hyperthermia enhances photodynamic therapy by regulation of HCP1 and ABCG2 expressions via high level ROS generation. Sci. Rep. 2019, 9, 1638. [Google Scholar] [CrossRef] [Green Version]
- Svedung Wettervik, T.M.; Engquist, H.; Lenell, S.; Howells, T.; Hillered, L.; Rostami, E.; Lewén, A.; Enblad, P. Systemic Hyperthermia in Traumatic Brain Injury—Relation to Intracranial Pressure Dynamics, Cerebral Energy Metabolism, and Clinical Outcome. J. Neurosurg. Anesthesiol. 2021, 33, 329–336. [Google Scholar] [CrossRef]
- Saxton, C. Effects of severe heat stress on respiration and metabolic rate in resting man. Aviat. Space Environ. Med. 1981, 52, 281–286. [Google Scholar]
- Mickley, G.A.; Cobb, B.L.; Farrell, S.T. Brain hyperthermia alters local cerebral glucose utilization: A comparison of hyperthermic agents. Int. J. Hyperth. 1997, 13, 99–114. [Google Scholar] [CrossRef]
- Nunneley, S.A.; Martin, C.C.; Slauson, J.W.; Hearon, C.M.; Nickerson, L.D.H.; Mason, P.A. Changes in regional cerebral metabolism during systemic hyperthermia in humans. J. Appl. Physiol. 2002, 92, 846–851. [Google Scholar] [CrossRef] [Green Version]
- Spiotta, A.M.; Stiefel, M.F.; Heuer, G.G.; Bloom, S.; Maloney-Wilensky, E.; Yang, W.; Grady, M.S.; Le Roux, P.D. Brain Hyperthermia After Traumatic Brain Injury Does Not Reduce Brain Oxygen. Neurosurgery 2008, 62, 864–872. [Google Scholar] [CrossRef]
- Schumacker, P.T.; Rowland, J.; Saltz, S.; Nelson, D.P.; Wood, L.D. Effects of hyperthermia and hypothermia on oxygen extraction by tissues during hypovolemia. J. Appl. Physiol. 1987, 63, 1246–1252. [Google Scholar] [CrossRef]
- Edvinsson, L.; MacKenzie, E.T.; McCulloch, J. Cerebral Blood Flow and Metabolism; Raven Press: New York, NY, USA, 1993. [Google Scholar]
- Crandall, C.G.; Gonzalez-Alonso, J. Cardiovascular function in the heat-stressed human. Acta Physiol. 2010, 199, 407–423. [Google Scholar] [CrossRef] [Green Version]
- Gross, P.M.; Harper, A.M.; Teasdale, G.M. Interaction of histamine with noradrenergic constrictory mechanisms in cat cerebral arteries and veins. Can. J. Physiol. Pharmacol. 1983, 61, 756–763. [Google Scholar] [CrossRef]
- Edvinsson, L. Sympathetic control of cerebral circulation. Trends Neurosci. 1982, 5, 425–429. [Google Scholar] [CrossRef]
- Watson, P.; Black, K.E.; Clark, S.C.; Maughan, R.J. Exercise in the heat: Effect of fluid ingestion on blood-brain barrier permeability. Med. Sci. Sport. Exerc. 2006, 38, 2118–2124. [Google Scholar] [CrossRef]
- Watson, P.; Shirreffs, S.M.; Maughan, R.J. Blood-brain barrier integrity may be threatened by exercise in a warm environment. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R1689–R1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, E.J.; Hanna-Jumma, S.; Carraretto, M.; Forni, L. The pathophysiological basis and consequences of fever. Crit. Care 2016, 20, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilpatrick, M.M.; Lowry, D.W.; Firlik, A.D.; Yonas, H.; Marion, D.W. Hyperthermia in the neurosurgical intensive care unit. Neurosurgery 2000, 47, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Weng, W.-J.; Yang, C.; Huang, X.-J.; Zhang, Y.-M.; Liu, J.-F.; Yao, J.-M.; Zi Zhang, Z.; Wu, X.; Mei, T.; Zhang, C.; et al. Effects of brain temperature on the outcome of patients with traumatic brain injury: A prospective observational study. J. Neurotrauma 2019, 36, 1168–1174. [Google Scholar] [CrossRef] [PubMed]
- Holtzclaw, B.J. The febrile response in critical care: State of the science. Heart Lung J. Crit. Care 1992, 21, 482–501. [Google Scholar]
- Bain, A.R.; Smith, K.J.; Lewis, N.C.; Foster, G.E.; Wildfong, K.W.; Willie, C.K.; Hartley, G.L.; Cheung, S.S.; Ainslie, P.N. Regional changes in brain blood flow during severe passive hyperthermia: Effects of PaCO2 and extracranial blood flow. J. Appl. Physiol. 2013, 115, 653–659. [Google Scholar] [CrossRef] [Green Version]
- Stocchetti, N.; Protti, A.; Lattuada, M.; Magnoni, S.; Longhi, L.; Ghisoni, L.; Egidi, M.; Zanier, E. Impact of pyrexia on neurochemistry and cerebral oxygenation after acute brain injury. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1135–1139. [Google Scholar] [CrossRef]
- Rossi, S.; Zanier, E.R.; Mauri, I.; Columbo, A.; Stocchetti, N. Brain temperature, body core temperature, and intracranial pressure in acute cerebral damage. J. Neurol. Neurosurg. Psychiatry 2001, 71, 448–454. [Google Scholar] [CrossRef] [Green Version]
- Nyholm, L.; Howells, T.; Lewén, A.; Hillered, L.; Enblad, P. The influence of hyperthermia on intracranial pressure, cerebral oximetry and cerebral metabolism in traumatic brain injury. Upsala J. Med. Sci. 2017, 122, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, B.; Normoyle, K.P.; Jackson, K.; Spitler, K.; Sharrock, M.F.; Miller, C.M.; Best, C.; Llano, D.; Du, R. Brain temperature and its fundamental properties: A review for clinical neuroscientists. Front. Neurosci. 2014, 8, 307. [Google Scholar] [CrossRef]
- Le Roux, P.; Menon, D.K.; Citerio, G.; Vespa, P.; Bader, M.K.; Brophy, G.M.; Diringer, M.N.; Stocchetti, N.; Videtta, W.; Armonda, R.; et al. Consensus summary statement of the international multidisciplinary consensus conference on multimodality monitoring in neurocritical care. Neurocritical Care 2014, 21, 1–26. [Google Scholar] [CrossRef]
- Rass, V.; Huber, L.; Ianosi, B.-A.; Kofler, M.; Lindner, A.; Picetti, E.; Ortolano, F.; Beer, R.; Rossi, S.; Smielewski, P.; et al. The Effect of Temperature Increases on Brain Tissue Oxygen Tension in Patients with Traumatic Brain Injury: A Collaborative European NeuroTrauma Effectiveness Research in Traumatic Brain Injury Substudy. Ther. Hypothermia Temp. Manag. 2020, 11, 122–131. [Google Scholar] [CrossRef]
- Kil, H.Y.; Zhang, J.; Piantadosi, C.A. Brain Temperature Alters Hydroxyl Radical Production during Cerebral Ischemia/Reperfusion in Rats. J. Cereb. Blood Flow Metab. 1996, 16, 100–106. [Google Scholar] [CrossRef] [Green Version]
- Dempsey, R.J.; Combs, D.J.; Maley, M.E.; Cowen, D.E.; Roy, M.W.; Donaldson, D.L. Moderate hypothermia reduces postischemic edema development and leukotriene production. Neurosurgery 1987, 21, 177–181. [Google Scholar] [CrossRef]
- Haba, K.; Ogawa, N.; Mizukawa, K.; Mori, A. Time course of changes in lipid peroxidation, pre-and postsynaptic cholinergic indices, NMDA receptor binding and neuronal death in the gerbil hippocampus following transient ischemia. Brain Res. 1991, 540, 116–122. [Google Scholar] [CrossRef]
- Wells, C.E. The Cerebral Circulation: The Clinical Significance of Current Concepts. Arch. Neurol. 1960, 3, 319–331. [Google Scholar] [CrossRef]
- Choi, H.A.; Badjatia, N.; Mayer, S.A. Hypothermia for acute brain injury—mechanisms and practical aspects. Nat. Rev. Neurol. 2012, 8, 214–222. [Google Scholar] [CrossRef]
- Dietrich, W.D.; Halley, M.; Valdes, I.; Busto, R. Interrelationships between increased vascular permeability and acute neuronal damage following temperature-controlled brain ischemia in rats. Acta Neuropathol. 1991, 81, 615–625. [Google Scholar] [CrossRef]
- Ginsberg, M.D.; Sternau, L.L.; Globus, M.Y.; Dietrich, W.D.; Busto, R. Therapeutic modulation of brain temperature: Relevance to ischemic brain injury. Cerebrovasc. Brain Metab. Rev. 1992, 4, 189–225. [Google Scholar]
- Kramer, R.S.; Sanders, A.P.; Lesage, A.M.; Woodhall, B.; Sealy, W.C. The effect of profound hypothermia on preservation of cerebral ATP content during circulatory arrest. J. Thorac. Cardiovasc. Surg. 1968, 56, 699–709. [Google Scholar] [CrossRef]
- Taft, W.C.; Yang, K.; Dixon, C.E.; Clifton, G.L.; Hayes, R.L. Hypothermia attenuates the loss of hippocampal microtubule-associated protein 2 (MAP2) following traumatic brain injury. J. Cereb. Blood Flow Metab. 1993, 13, 796–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dede, S.; Deger, Y.; Meral, I. Effect of Short-term Hypothermia on Lipid Peroxidation and Antioxidant Enzyme Activity in Rats. J. Vet. Med. Ser. A 2002, 49, 286–288. [Google Scholar] [CrossRef] [PubMed]
- Truettner, J.S.; Bramlett, H.M.; Dietrich, W.D. Posttraumatic therapeutic hypothermia alters microglial and macrophage polarization toward a beneficial phenotype. J. Cereb. Blood Flow Metab. 2017, 37, 2952–2962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erecinska, M.; Thoresen, M.; Silver, I.A. Effects of Hypothermia on Energy Metabolism in Mammalian Central Nervous System. J. Cereb. Blood Flow Metab. 2003, 23, 513–530. [Google Scholar] [CrossRef] [PubMed]
- Walter, B.; Bauer, R.; Kuhnen, G.; Fritz, H.; Zwiener, U. Coupling of cerebral blood flow and oxygen metabolism in infant pigs during selective brain hypothermia. J. Cereb. Blood Flow Metab. 2000, 20, 1215–1224. [Google Scholar] [CrossRef]
- Jensen, A.; Garnier, Y.; Berger, R. Dynamics of fetal circulatory responses to hypoxia and asphyxia. Eur. J. Obstet. Gynecol. Reprod. Biol. 1999, 84, 155–172. [Google Scholar] [CrossRef]
- Chihara, H.; Blood, A.B.; Hunter, C.J.; Power, G.G. Effect of Mild Hypothermia and Hypoxia on Blood Flow and Oxygen Consumption of the Fetal Sheep Brain. Pediatr. Res. 2003, 54, 665–671. [Google Scholar] [CrossRef] [Green Version]
- Hashem, M.; Zhang, Q.; Wu, Y.; Johnson, T.W.; Dunn, J.F. Using a multimodal near-infrared spectroscopy and MRI to quantify gray matter metabolic rate for oxygen: A hypothermia validation study. NeuroImage 2020, 206, 116315. [Google Scholar] [CrossRef]
- Pichler, G.; Baumgartner, S.; Biermayr, M.; Dempsey, E.; Fuchs, H.; Goos, T.G.; Lista, G.; Lorenz, L.; Karpinski, L.; Mitra, S.; et al. Cerebral regional tissue Oxygen Saturation to Guide Oxygen Delivery in preterm neonates during immediate transition after birth (COSGOD III): An investigator-initiated, randomized, multi-center, multi-national, clinical trial on additional cerebral tissue oxygen saturation monitoring combined with defined treatment guidelines versus standard monitoring and treatment as usual in premature infants during immediate transition: Study protocol for a randomized controlled trial. Trials 2019, 20, 178. [Google Scholar] [CrossRef]
- Laurikkala, J.; Aneman, A.; Peng, A.; Reinikainen, M.; Pham, P.; Jakkula, P.; Hästbacka, J.; Wilkman, E.; Loisa, P.; Toppila, J.; et al. Association of deranged cerebrovascular reactivity with brain injury following cardiac arrest: A post-hoc analysis of the COMACARE trial. Crit. Care 2021, 25, 350. [Google Scholar] [CrossRef]
- Clifton, G.L.; Allen, S.; Barrodale, P.; Plenger, P.; Berry, J.; Koch, S.; Fletcher, J.; Hayes, R.L.; Choi, S.C. A Phase II Study of Moderate Hypothermia in Severe Brain Injury. J. Neurotrauma 1993, 10, 263–271. [Google Scholar] [CrossRef]
- Rösli, D.; Schnüriger, B.; Candinas, D.; Haltmeier, T. The Impact of Accidental Hypothermia on Mortality in Trauma Patients Overall and Patients with Traumatic Brain Injury Specifically: A Systematic Review and Meta-Analysis. World J. Surg. 2020, 44, 4106–4117. [Google Scholar] [CrossRef]
- Wu, X.; Tao, Y.; Marsons, L.; Dee, P.; Yu, D.; Guan, Y.; Zhou, X. The effectiveness of early prophylactic hypothermia in adult patients with traumatic brain injury: A systematic review and meta-analysis. Aust. Crit. Care 2021, 34, 83–91. [Google Scholar] [CrossRef]
- Tokutomi, T.; Morimoto, K.; Miyagi, T.; Yamaguchi, S.; Ishikawa, K.; Shigemori, M. Optimal Temperature For The Management Of Severe Traumatic Brain Injury: Effect Of Hypothermia On Intracranial Pressure, Systemic And Intracranial Hemodynamics, And Metabolism. Neurosurgery 2007, 61 (Suppl. 1), 102–112. [Google Scholar] [CrossRef]
- Ghosh, A.; Highton, D.; Kolyva, C.; Tachtsidis, I.; Elwell, C.E.; Smith, M. Hyperoxia results in increased aerobic metabolism following acute brain injury. J. Cereb. Blood Flow Metab. 2017, 37, 2910–2920. [Google Scholar] [CrossRef]
- Yang, D.; Ma, L.; Wang, P.; Yang, D.; Zhang, Y.; Zhao, X.; Lv, J.; Zhang, J.; Zhang, Z.; Gao, F. Normobaric oxygen inhibits AQP4 and NHE1 expression in experimental focal ischemic stroke. Int. J. Mol. Med. 2019, 43, 1193–1202. [Google Scholar] [CrossRef]
- Liang, J.; Qi, Z.; Liu, W.; Wang, P.; Shi, W.; Dong, W.; Ji, X.; Luo, Y.; Liu, K.J. Normobaric Hyperoxia Slows Blood–Brain Barrier Damage and Expands the Therapeutic Time Window for Tissue-Type Plasminogen Activator Treatment in Cerebral Ischemia. Stroke 2015, 46, 1344–1351. [Google Scholar] [CrossRef] [Green Version]
- Braswell, C.; Crowe, D.T. Hyperbaric oxygen therapy. Compend. Contin. Educ. Vet. 2012, 34, E1–E6. [Google Scholar]
- Sanchez, E.C. Mechanisms of action of hyperbaric oxygenation in stroke: A review. Crit. Care Nurs. Q. 2013, 36, 290–298. [Google Scholar] [CrossRef]
- Liang, F.; Sun, L.; Yang, J.; Liu, X.-H.; Zhang, J.; Zhu, W.-Q.; Yang, L.; Nan, D. The effect of different atmosphere absolute hyperbaric oxygen on the expression of extracellular histones after traumatic brain injury in rats. Cell Stress Chaperones 2020, 25, 1013–1024. [Google Scholar] [CrossRef]
- Xu, Z.; Huang, Y.; Mao, P.; Zhang, J.; Li, Y. Sepsis and ARDS: The dark side of histones. Mediat. Inflamm. 2015, 2015, 205054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palzur, E.; Vlodavsky, E.; Mulla, H.; Arieli, R.; Feinsod, M.; Soustiel, J.F. Hyperbaric oxygen therapy for reduction of secondary brain damage in head injury: An animal model of brain contusion. J. Neurotrauma 2004, 21, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palzur, E.; Zaaroor, M.; Vlodavsky, E.; Milman, F.; Soustiel, J.F. Neuroprotective effect of hyperbaric oxygen therapy in brain injury is mediated by preservation of mitochondrial membrane properties. Brain Res. 2008, 1221, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Rockswold, G.L.; Ford, S.E.; Anderson, D.C.; Bergman, T.A.; Sherman, R.E. Results of a prospective randomized trial for treatment of severely brain-injured patients with hyperbaric oxygen. J. Neurosurg. 1992, 76, 929–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rockswold, S.B.; Rockswold, G.L.; Zaun, D.A.; Liu, J. A prospective, randomized Phase II clinical trial to evaluate the effect of combined hyperbaric and normobaric hyperoxia on cerebral metabolism, intracranial pressure, oxygen toxicity, and clinical outcome in severe traumatic brain injury. J. Neurosurg. 2013, 118, 1317–1328. [Google Scholar] [CrossRef] [Green Version]
- Harch, P.G.; Andrews, S.R.; Rowe, C.J.; Lischka, J.R.; Townsend, M.H.; Yu, Q.; E Mercante, D. Hyperbaric oxygen therapy for mild traumatic brain injury persistent postconcussion syndrome: A randomized controlled trial. Med. Gas Res. 2020, 10, 8–20. [Google Scholar] [CrossRef]
- Amir, H.; Shai, E. The Hyperoxic-Hypoxic Paradox. Biomolecules 2020, 10, 958. [Google Scholar] [CrossRef]
- Annoni, F.; Peluso, L.; Bogossian, E.G.; Creteur, J.; Zanier, E.; Taccone, F. Brain Protection after Anoxic Brain Injury: Is Lactate Supplementation Helpful? Cells 2021, 10, 1714. [Google Scholar] [CrossRef]
- Phillis, J.W.; Song, D.; Guyot, L.L.; O’Regan, M.H. Lactate reduces amino acid release and fuels recovery of function in the ischemic brain. Neurosci. Lett. 1999, 272, 195–198. [Google Scholar] [CrossRef]
- Berthet, C.; Lei, H.; Thevenet, J.; Gruetter, R.; Magistretti, P.J.; Hirt, L. Neuroprotective role of lactate after cerebral ischemia. J. Cereb. Blood Flow Metab. 2009, 29, 1780–1789. [Google Scholar] [CrossRef]
- Horn, T.; Klein, J. Neuroprotective effects of lactate in brain ischemia: Dependence on anesthetic drugs. Neurochem. Int. 2013, 62, 251–257. [Google Scholar] [CrossRef]
- Berthet, C.; Castillo, X.; Magistretti, P.J.; Hirt, L. New evidence of neuroprotection by lactate after transient focal cerebral ischaemia: Extended benefit after intracerebroventricular injection and efficacy of intravenous administration. Cerebrovasc. Dis. 2012, 34, 329–335. [Google Scholar] [CrossRef]
- Ichai, C.; Armando, G.; Orban, J.-C.; Berthier, F.; Rami, L.; Samat-Long, C.; Grimaud, D.; Leverve, X. Sodium lactate versus mannitol in the treatment of intracranial hypertensive episodes in severe traumatic brain-injured patients. Intensiv. Care Med. 2009, 35, 471–479. [Google Scholar] [CrossRef]
- Bouzat, P.; Sala, N.; Suys, T.; Zerlauth, J.-B.; Marques-Vidal, P.; Feihl, F.; Bloch, J.; Messerer, M.; Levivier, M.; Meuli, R.; et al. Cerebral metabolic effects of exogenous lactate supplementation on the injured human brain. Intensiv. Care Med. 2014, 40, 412–421. [Google Scholar] [CrossRef] [Green Version]
- Krep, H.; Breil, M.; Sinn, D.; Hagendorff, A.; Hoeft, A.; Fischer, M. Effects of hypertonic versus isotonic infusion therapy on regional cerebral blood flow after experimental cardiac arrest cardiopulmonary resuscitation in pigs. Resuscitation 2004, 63, 73–83. [Google Scholar] [CrossRef]
- Ros, J.; Pecinska, N.; Alessandri, B.; Landolt, H.; Fillenz, M. Lactate reduces glutamate-induced neurotoxicity in rat cortex. J. Neurosci. Res. 2001, 66, 790–794. [Google Scholar] [CrossRef]
- Sørensen, A.T.; Ledri, M.; Melis, M.; Nikitidou Ledri, L.; Andersson, M.; Kokaia, M. Altered Chloride Homeo-stasis Decreases the Action Potential Threshold and Increases Hyperexcitability in Hippocampal Neu-rons. eNeuro 2018, 4. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Hypoxia | Normoxia | Hyperoxia | |
| Oxidative stress | ↑↑ | ↑ | ↑↑↑ |
| Hypoxia-inducible factor (HIF) | ↑↑ | ↑ | ↑↑ |
| Protein kinase B (Akt) | ↑↑ | ↑ | ↑↑ |
| Extracellular signal-regulated kinase (ERK) | ↑↑ | ↑ | ↑↑ |
| Brain-derived neurotrophic factor (BDNF) | ↑↑ | ↑ | ↑↑ |
| Erythropoietin (Epo) | ↑↑ | ↑ | ↑↑ |
| Neuroglobin (Ngb) | ↑↑ | ↑ | ↑↑ |
| Nitric oxide (NO) | ↑↑ | ↑ | ↑↑ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siwicka-Gieroba, D.; Robba, C.; Gołacki, J.; Badenes, R.; Dabrowski, W. Cerebral Oxygen Delivery and Consumption in Brain-Injured Patients. J. Pers. Med. 2022, 12, 1763. https://doi.org/10.3390/jpm12111763
Siwicka-Gieroba D, Robba C, Gołacki J, Badenes R, Dabrowski W. Cerebral Oxygen Delivery and Consumption in Brain-Injured Patients. Journal of Personalized Medicine. 2022; 12(11):1763. https://doi.org/10.3390/jpm12111763
Chicago/Turabian StyleSiwicka-Gieroba, Dorota, Chiara Robba, Jakub Gołacki, Rafael Badenes, and Wojciech Dabrowski. 2022. "Cerebral Oxygen Delivery and Consumption in Brain-Injured Patients" Journal of Personalized Medicine 12, no. 11: 1763. https://doi.org/10.3390/jpm12111763