
Status of ALS Treatment, Insights into Therapeutic Challenges and Dilemmas
Abstract
:1. Introduction
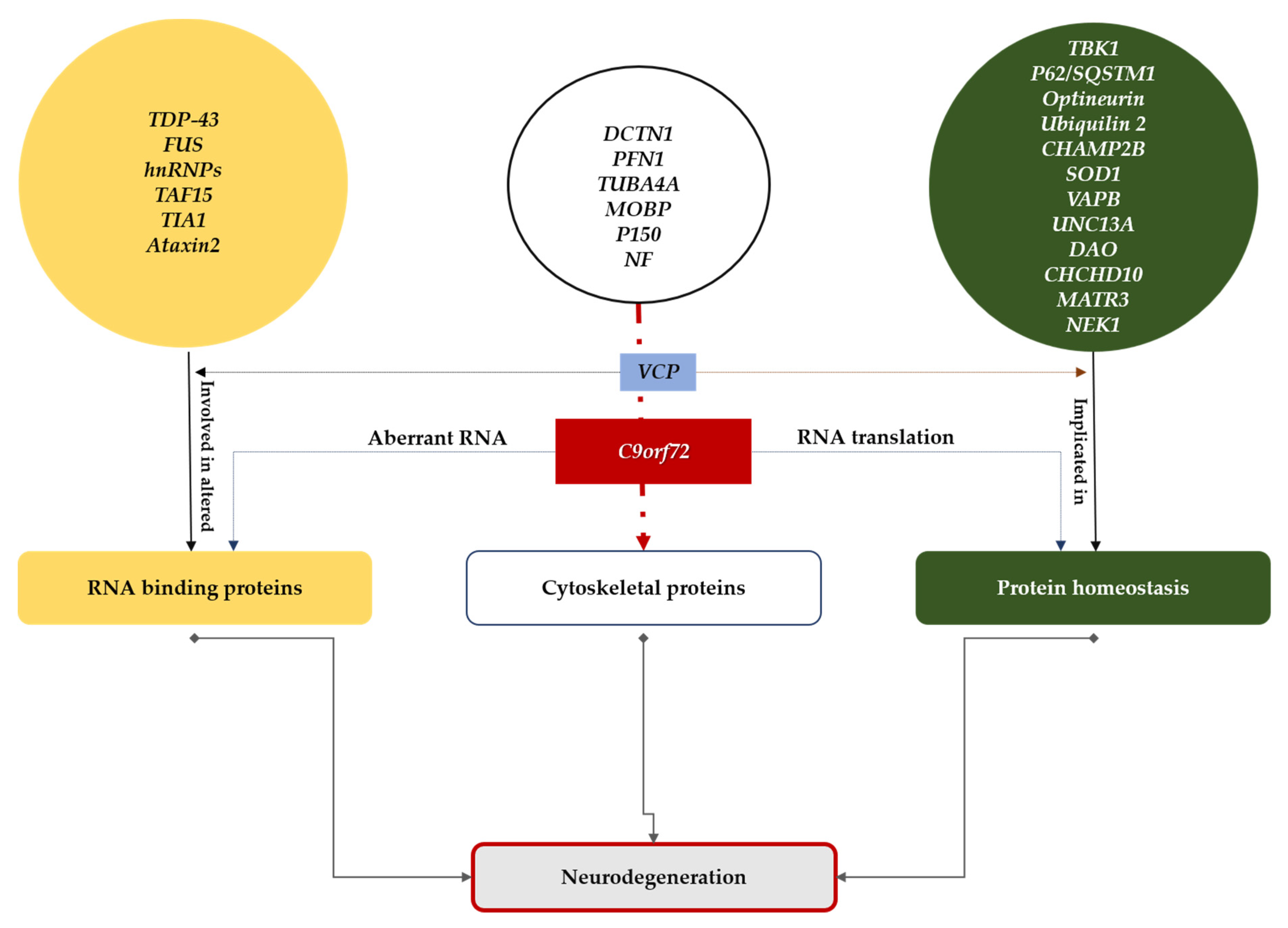
2. Some Aspects of ALS Disease Mechanisms Acted upon by Therapeutic Strategies
3. Therapeutic Approaches
3.1. Pharmacologic Approaches
3.2. Gene and Cell Therapy Approaches
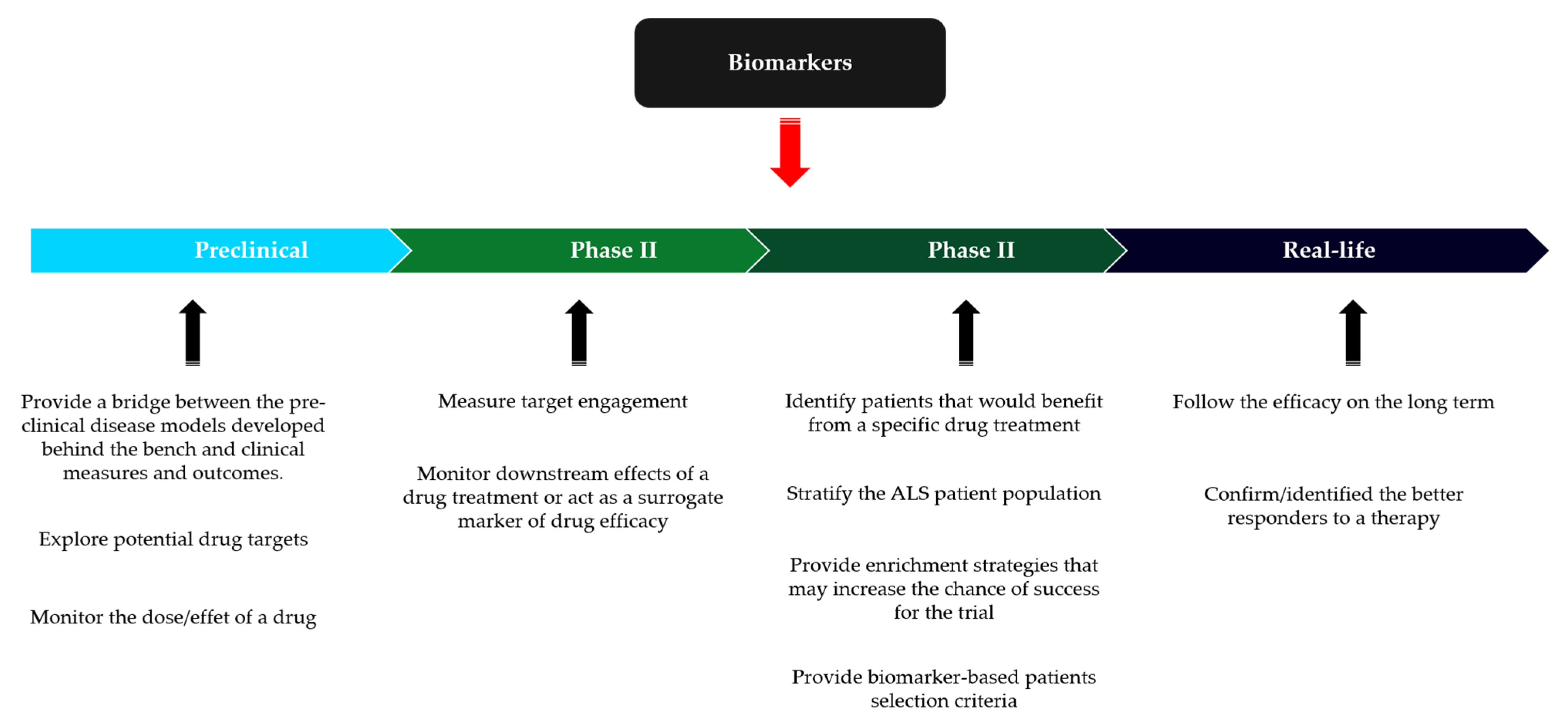
4. Preclinical and Clinical Development
4.1. Improve Preclinical Models
4.2. Design of Clinical Trial in ALS
4.3. Economic and Ethical Realities
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Bensimon, G.; Lacomblez, L.; Meininger, V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N. Engl. J. Med. 1994, 330, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Aoki, M.; Tsuji, S.; Itoyama, Y.; Sobue, G.; Togo, M.; Hamada, C.; Tanaka, M.; Akimoto, M.; Nakamura, K.; et al. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017, 16, 505–512. [Google Scholar] [CrossRef]
- Traynor, B.J.; Alexander, M.; Corr, B.; Frost, E.; Hardiman, O. Effect of a multidisciplinary amyotrophic lateral sclerosis (ALS) clinic on ALS survival: A population based study, 1996-2000. J. Neurol. Neurosurg. Psychiatry 2003, 74, 1258–1261. [Google Scholar] [CrossRef] [PubMed]
- Radunovic, A.; Annane, D.; Rafiq, M.K.; Brassington, R.; Mustfa, N. Mechanical ventilation for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst. Rev. 2017, 10, Cd004427. [Google Scholar] [CrossRef]
- Zhao, M.; Kim, J.R.; van Bruggen, R.; Park, J. RNA-Binding Proteins in Amyotrophic Lateral Sclerosis. Mol. Cells 2018, 41, 818–829. [Google Scholar] [CrossRef]
- Le Gall, L.; Anakor, E.; Connolly, O.; Vijayakumar, U.G.; Duddy, W.J.; Duguez, S. Molecular and Cellular Mechanisms Affected in ALS. J. Pers. Med. 2020, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- Hulisz, D. Amyotrophic lateral sclerosis: Disease state overview. Am. J. Manag. Care 2018, 24, S320–S326. [Google Scholar] [PubMed]
- Rosen, D.R.; Sapp, P.; O’Regan, J.; McKenna-Yasek, D.; Schlumpf, K.S.; Haines, J.L.; Gusella, J.F.; Horvitz, H.R.; Brown, R.H., Jr. Genetic linkage analysis of familial amyotrophic lateral sclerosis using human chromosome 21 microsatellite DNA markers. Am. J. Med. Genet. 1994, 51, 61–69. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed]
- van Blitterswijk, M.; DeJesus-Hernandez, M.; Rademakers, R. How do C9ORF72 repeat expansions cause amyotrophic lateral sclerosis and frontotemporal dementia: Can we learn from other noncoding repeat expansion disorders? Curr. Opin. Neurol. 2012, 25, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.S.; Holzbaur, E.L.F. Autophagy and mitophagy in ALS. Neurobiol. Dis. 2019, 122, 35–40. [Google Scholar] [CrossRef]
- Burk, K.; Pasterkamp, R.J. Disrupted neuronal trafficking in amyotrophic lateral sclerosis. Acta Neuropathol. 2019, 137, 859–877. [Google Scholar] [CrossRef]
- Kaur, B.; Bhat, A.; Chakraborty, R.; Adlakha, K.; Sengupta, S.; Roy, S.; Chakraborty, K. Proteomic profile of 4-PBA treated human neuronal cells during ER stress. Mol. Omics 2018, 14, 53–63. [Google Scholar] [CrossRef]
- Suaud, L.; Miller, K.; Panichelli, A.E.; Randell, R.L.; Marando, C.M.; Rubenstein, R.C. 4-Phenylbutyrate stimulates Hsp70 expression through the Elp2 component of elongator and STAT-3 in cystic fibrosis epithelial cells. J. Biol. Chem. 2011, 286, 45083–45092. [Google Scholar] [CrossRef]
- Rodrigues, C.M.; Solá, S.; Sharpe, J.C.; Moura, J.J.; Steer, C.J. Tauroursodeoxycholic acid prevents Bax-induced membrane perturbation and cytochrome C release in isolated mitochondria. Biochemistry 2003, 42, 3070–3080. [Google Scholar] [CrossRef]
- Paganoni, S.; Macklin, E.A.; Hendrix, S.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; Owegi, M.A.; Quick, A.; et al. Trial of Sodium Phenylbutyrate-Taurursodiol for Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2020, 383, 919–930. [Google Scholar] [CrossRef]
- Mandrioli, J.; Crippa, V.; Cereda, C.; Bonetto, V.; Zucchi, E.; Gessani, A.; Ceroni, M.; Chio, A.; D’Amico, R.; Monsurrò, M.R.; et al. Proteostasis and ALS: Protocol for a phase II, randomised, double-blind, placebo-controlled, multicentre clinical trial for colchicine in ALS (Co-ALS). BMJ Open 2019, 9, e028486. [Google Scholar] [CrossRef] [PubMed]
- Mandrioli, J.; D’Amico, R.; Zucchi, E.; Gessani, A.; Fini, N.; Fasano, A.; Caponnetto, C.; Chiò, A.; Dalla Bella, E.; Lunetta, C.; et al. Rapamycin treatment for amyotrophic lateral sclerosis: Protocol for a phase II randomized, double-blind, placebo-controlled, multicenter, clinical trial (RAP-ALS trial). Medicine 2018, 97, e11119. [Google Scholar] [CrossRef] [PubMed]
- Devos, D.; Cabantchik, Z.I.; Moreau, C.; Danel, V.; Mahoney-Sanchez, L.; Bouchaoui, H.; Gouel, F.; Rolland, A.S.; Duce, J.A.; Devedjian, J.C. Conservative iron chelation for neurodegenerative diseases such as Parkinson’s disease and amyotrophic lateral sclerosis. J. Neural Transm. 2020, 127, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Li, M.X.; Mercier, P.; Hartman, J.J.; Sykes, B.D. Structural Basis of Tirasemtiv Activation of Fast Skeletal Muscle. J. Med. Chem. 2021, 64, 3026–3034. [Google Scholar] [CrossRef] [PubMed]
- Shefner, J.M.; Cudkowicz, M.E.; Hardiman, O.; Cockcroft, B.M.; Lee, J.H.; Malik, F.I.; Meng, L.; Rudnicki, S.A.; Wolff, A.A.; Andrews, J.A. A phase III trial of tirasemtiv as a potential treatment for amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 584–594. [Google Scholar] [CrossRef]
- Camu, W.; Mickunas, M.; Veyrune, J.L.; Payan, C.; Garlanda, C.; Locati, M.; Juntas-Morales, R.; Pageot, N.; Malaspina, A.; Andreasson, U.; et al. Repeated 5-day cycles of low dose aldesleukin in amyotrophic lateral sclerosis (IMODALS): A phase 2a randomised, double-blind, placebo-controlled trial. EBioMedicine 2020, 59, 102844. [Google Scholar] [CrossRef]
- Mora, J.S.; Bradley, W.G.; Chaverri, D.; Hernández-Barral, M.; Mascias, J.; Gamez, J.; Gargiulo-Monachelli, G.M.; Moussy, A.; Mansfield, C.D.; Hermine, O.; et al. Long-term survival analysis of masitinib in amyotrophic lateral sclerosis. Ther. Adv. Neurol. Disord. 2021, 14, 17562864211030365. [Google Scholar] [CrossRef]
- Gibson, L.C.; Hastings, S.F.; McPhee, I.; Clayton, R.A.; Darroch, C.E.; Mackenzie, A.; Mackenzie, F.L.; Nagasawa, M.; Stevens, P.A.; Mackenzie, S.J. The inhibitory profile of Ibudilast against the human phosphodiesterase enzyme family. Eur. J. Pharmacol. 2006, 538, 39–42. [Google Scholar] [CrossRef]
- Babu, S.; Hightower, B.G.; Chan, J.; Zürcher, N.R.; Kivisäkk, P.; Tseng, C.J.; Sanders, D.L.; Robichaud, A.; Banno, H.; Evora, A.; et al. Ibudilast (MN-166) in amyotrophic lateral sclerosis-an open label, safety and pharmacodynamic trial. NeuroImage Clin. 2021, 30, 102672. [Google Scholar] [CrossRef]
- Oskarsson, B.; Maragakis, N.; Bedlack, R.S.; Goyal, N.; Meyer, J.A.; Genge, A.; Bodkin, C.; Maiser, S.; Staff, N.; Zinman, L.; et al. MN-166 (ibudilast) in amyotrophic lateral sclerosis in a Phase IIb/III study: COMBAT-ALS study design. Neurodegener. Dis. Manag. 2021, 11, 431–443. [Google Scholar] [CrossRef]
- Lingor, P.; Weber, M.; Camu, W.; Friede, T.; Hilgers, R.; Leha, A.; Neuwirth, C.; Günther, R.; Benatar, M.; Kuzma-Kozakiewicz, M.; et al. ROCK-ALS: Protocol for a Randomized, Placebo-Controlled, Double-Blind Phase IIa Trial of Safety, Tolerability and Efficacy of the Rho Kinase (ROCK) Inhibitor Fasudil in Amyotrophic Lateral Sclerosis. Front. Neurol. 2019, 10, 293. [Google Scholar] [CrossRef] [PubMed]
- Astrazeneca. Update on CHAMPION-ALS Phase III Trial of Ultomiris in Amyotrophic Lateral Sclerosis. Available online: https://www.astrazeneca.com/media-centre/press-releases/2021/update-on-ultomiris-phase-iii-als-trial.html (accessed on 28 February 2022).
- Hospital, M.G. HEALEY ALS Platform Trial Update: Zilucoplan Arm Stopped Early for Futility. Available online: https://www.massgeneral.org/news/press-release/healey-als-platform-trial-update-ilucoplan-arm-stopped-early-for-futility (accessed on 3 March 2022).
- Milligan, C.; Atassi, N.; Babu, S.; Barohn, R.J.; Caress, J.B.; Cudkowicz, M.E.; Evora, A.; Hawkins, G.A.; Wosiski-Kuhn, M.; Macklin, E.A.; et al. Tocilizumab is safe and tolerable and reduces C-reactive protein concentrations in the plasma and cerebrospinal fluid of ALS patients. Muscle Nerve 2021, 64, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Biogen Announces Topline Results from the Tofersen Phase 3 Study and its Open-Label Extension in SOD1-ALS. Available online: https://www.globenewswire.com/fr/news-release/2021/10/17/2315291/0/en/Biogen-Announces-Topline-Results-from-the-Tofersen-Phase-3-Study-and-its-Open-Label-Extension-in-SOD1-ALS.html (accessed on 1 March 2022).
- Biogen. Biogen and Ionis Announce Topline Phase 1 Study Results of Investigational Drug in C9orf72 Amyotrophic Lateral Sclerosis. Available online: https://investors.biogen.com/news-releases/news-release-details/biogen-and-ionis-announce-topline-phase-1-study-results (accessed on 4 March 2022).
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Kalmar, B.; Greensmith, L. Cellular Chaperones As Therapeutic Targets in ALS to Restore Protein Homeostasis and Improve Cellular Function. Front. Mol. Neurosci. 2017, 10, 251. [Google Scholar] [CrossRef]
- Trippier, P.C.; Zhao, K.T.; Fox, S.G.; Schiefer, I.T.; Benmohamed, R.; Moran, J.; Kirsch, D.R.; Morimoto, R.I.; Silverman, R.B. Proteasome activation is a mechanism for pyrazolone small molecules displaying therapeutic potential in amyotrophic lateral sclerosis. ACS Chem. Neurosci. 2014, 5, 823–829. [Google Scholar] [CrossRef]
- Kim, H.J.; Taylor, J.P. Lost in Transportation: Nucleocytoplasmic Transport Defects in ALS and Other Neurodegenerative Diseases. Neuron 2017, 96, 285–297. [Google Scholar] [CrossRef]
- Chou, C.C.; Zhang, Y.; Umoh, M.E.; Vaughan, S.W.; Lorenzini, I.; Liu, F.; Sayegh, M.; Donlin-Asp, P.G.; Chen, Y.H.; Duong, D.M.; et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat. Neurosci. 2018, 21, 228–239. [Google Scholar] [CrossRef]
- Yamanaka, K.; Chun, S.J.; Boillee, S.; Fujimori-Tonou, N.; Yamashita, H.; Gutmann, D.H.; Takahashi, R.; Misawa, H.; Cleveland, D.W. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 2008, 11, 251–253. [Google Scholar] [CrossRef]
- Boillée, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and progression in inherited ALS determined by motor neurons and microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Van Kammen, M.; Levey, A.I.; Martin, L.J.; Kuncl, R.W. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1995, 38, 73–84. [Google Scholar] [CrossRef]
- Ferraiuolo, L.; Meyer, K.; Sherwood, T.W.; Vick, J.; Likhite, S.; Frakes, A.; Miranda, C.J.; Braun, L.; Heath, P.R.; Pineda, R.; et al. Oligodendrocytes contribute to motor neuron death in ALS via SOD1-dependent mechanism. Proc. Natl. Acad. Sci. USA 2016, 113, E6496–E6505. [Google Scholar] [CrossRef] [PubMed]
- Klivenyi, P.; Kiaei, M.; Gardian, G.; Calingasan, N.Y.; Beal, M.F. Additive neuroprotective effects of creatine and cyclooxygenase 2 inhibitors in a transgenic mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2004, 88, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Pompl, P.N.; Ho, L.; Bianchi, M.; McManus, T.; Qin, W.; Pasinetti, G.M. A therapeutic role for cyclooxygenase-2 inhibitors in a transgenic mouse model of amyotrophic lateral sclerosis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 725–727. [Google Scholar] [CrossRef] [PubMed]
- Kriz, J.; Nguyen, M.D.; Julien, J.P. Minocycline slows disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2002, 10, 268–278. [Google Scholar] [CrossRef]
- Boillée, S.; Vande Velde, C.; Cleveland, D.W. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron 2006, 52, 39–59. [Google Scholar] [CrossRef]
- Lee, J.; Hyeon, S.J.; Im, H.; Ryu, H.; Kim, Y.; Ryu, H. Astrocytes and Microglia as Non-cell Autonomous Players in the Pathogenesis of ALS. Exp. Neurobiol. 2016, 25, 233–240. [Google Scholar] [CrossRef]
- Clarke, B.E.; Patani, R. The microglial component of amyotrophic lateral sclerosis. Brain A J. Neurol. 2020, 143, 3526–3539. [Google Scholar] [CrossRef]
- Appel, S.H.; Beers, D.R.; Zhao, W. Amyotrophic lateral sclerosis is a systemic disease: Peripheral contributions to inflammation-mediated neurodegeneration. Curr. Opin. Neurol. 2021, 34, 765–772. [Google Scholar] [CrossRef]
- Beers, D.R.; Appel, S.H. Immune dysregulation in amyotrophic lateral sclerosis: Mechanisms and emerging therapies. Lancet Neurol. 2019, 18, 211–220. [Google Scholar] [CrossRef]
- Wei, Q.Q.; Hou, Y.B.; Zhang, L.Y.; Ou, R.W.; Cao, B.; Chen, Y.P.; Shang, H.F. Neutrophil-to-lymphocyte ratio in sporadic amyotrophic lateral sclerosis. Neural Regen. Res. 2022, 17, 875–880. [Google Scholar] [CrossRef]
- Liu, J.; Wang, F. Role of Neuroinflammation in Amyotrophic Lateral Sclerosis: Cellular Mechanisms and Therapeutic Implications. Front. Immunol. 2017, 8, 1005. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, S.; Garbelli, S.; Pasini, A.; Alimonti, D.; Perotti, C.; Melazzini, M.; Bendotti, C.; Mora, G. Immune system alterations in sporadic amyotrophic lateral sclerosis patients suggest an ongoing neuroinflammatory process. J. Neuroimmunol. 2009, 210, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Siddiqui, S.; Gabriely, G.; Lanser, A.J.; Dake, B.; Murugaiyan, G.; Doykan, C.E.; Wu, P.M.; Gali, R.R.; Iyer, L.K.; et al. Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J. Clin. Investig. 2012, 122, 3063–3087. [Google Scholar] [CrossRef] [PubMed]
- Zondler, L.; Müller, K.; Khalaji, S.; Bliederhäuser, C.; Ruf, W.P.; Grozdanov, V.; Thiemann, M.; Fundel-Clemes, K.; Freischmidt, A.; Holzmann, K.; et al. Peripheral monocytes are functionally altered and invade the CNS in ALS patients. Acta Neuropathol. 2016, 132, 391–411. [Google Scholar] [CrossRef]
- Sheean, R.K.; McKay, F.C.; Cretney, E.; Bye, C.R.; Perera, N.D.; Tomas, D.; Weston, R.A.; Scheller, K.J.; Djouma, E.; Menon, P.; et al. Association of Regulatory T-Cell Expansion With Progression of Amyotrophic Lateral Sclerosis: A Study of Humans and a Transgenic Mouse Model. JAMA Neurol. 2018, 75, 681–689. [Google Scholar] [CrossRef]
- Hooten, K.G.; Beers, D.R.; Zhao, W.; Appel, S.H. Protective and Toxic Neuroinflammation in Amyotrophic Lateral Sclerosis. Neurother. J. Am. Soc. Exp. NeuroTherapeutics 2015, 12, 364–375. [Google Scholar] [CrossRef]
- Verma, S.; Khurana, S.; Vats, A.; Sahu, B.; Ganguly, N.K.; Chakraborti, P.; Gourie-Devi, M.; Taneja, V. Neuromuscular Junction Dysfunction in Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2022, 59, 1502–1527. [Google Scholar] [CrossRef]
- Tarantino, N.; Canfora, I.; Camerino, G.M.; Pierno, S. Therapeutic Targets in Amyotrophic Lateral Sclerosis: Focus on Ion Channels and Skeletal Muscle. Cells 2022, 11, 415. [Google Scholar] [CrossRef]
- Pradat, P.F.; Dubourg, O.; de Tapia, M.; di Scala, F.; Dupuis, L.; Lenglet, T.; Bruneteau, G.; Salachas, F.; Lacomblez, L.; Corvol, J.C.; et al. Muscle gene expression is a marker of amyotrophic lateral sclerosis severity. Neurodegener. Dis. 2012, 9, 38–52. [Google Scholar] [CrossRef]
- Pradat, P.F.; Bruneteau, G.; Gonzalez de Aguilar, J.L.; Dupuis, L.; Jokic, N.; Salachas, F.; Le Forestier, N.; Echaniz-Laguna, A.; Dubourg, O.; Hauw, J.J.; et al. Muscle Nogo-A expression is a prognostic marker in lower motor neuron syndromes. Ann. Neurol. 2007, 62, 15–20. [Google Scholar] [CrossRef]
- Pradat, P.F.; Barani, A.; Wanschitz, J.; Dubourg, O.; Lombès, A.; Bigot, A.; Mouly, V.; Bruneteau, G.; Salachas, F.; Lenglet, T.; et al. Abnormalities of satellite cells function in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Off. Publ. World Fed. Neurol. Res. Group Mot. Neuron Dis. 2011, 12, 264–271. [Google Scholar] [CrossRef]
- Lunetta, C.; Serafini, M.; Prelle, A.; Magni, P.; Dozio, E.; Ruscica, M.; Sassone, J.; Colciago, C.; Moggio, M.; Corbo, M.; et al. Impaired expression of insulin-like growth factor-1 system in skeletal muscle of amyotrophic lateral sclerosis patients. Muscle Nerve 2012, 45, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, L.; Duddy, W.J.; Martinat, C.; Mariot, V.; Connolly, O.; Milla, V.; Anakor, E.; Ouandaogo, Z.G.; Millecamps, S.; Lainé, J.; et al. Muscle cells of sporadic amyotrophic lateral sclerosis patients secrete neurotoxic vesicles. J. Cachexia Sarcopenia Muscle 2022, 13, 1385–1402. [Google Scholar] [CrossRef]
- Scaricamazza, S.; Salvatori, I.; Giacovazzo, G.; Loeffler, J.P.; Renè, F.; Rosina, M.; Quessada, C.; Proietti, D.; Heil, C.; Rossi, S.; et al. Skeletal-Muscle Metabolic Reprogramming in ALS-SOD1(G93A) Mice Predates Disease Onset and Is A Promising Therapeutic Target. iScience 2020, 23, 101087. [Google Scholar] [CrossRef]
- Li, J.; Fredericks, M.; Cannell, M.; Wang, K.; Sako, D.; Maguire, M.C.; Grenha, R.; Liharska, K.; Krishnan, L.; Bloom, T.; et al. ActRIIB:ALK4-Fc alleviates muscle dysfunction and comorbidities in murine models of neuromuscular disorders. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Cappella, M.; Pradat, P.F.; Querin, G.; Biferi, M.G. Beyond the Traditional Clinical Trials for Amyotrophic Lateral Sclerosis and The Future Impact of Gene Therapy. J. Neuromuscul. Dis. 2021, 8, 25–38. [Google Scholar] [CrossRef]
- Miller, T.; Cudkowicz, M.; Shaw, P.J.; Andersen, P.M.; Atassi, N.; Bucelli, R.C.; Genge, A.; Glass, J.; Ladha, S.; Ludolph, A.L.; et al. Phase 1-2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2020, 383, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Westlund, R. Pre-Symptomatic ALS Clinical Trial Paves the Way for Early Intervention Strategies in Neurodegenerative Diseases. Available online: https://physician-news.umiamihealth.org/pre-symptomatic-als-clinical-trial-paves-the-way-for-early-intervention-strategies-in-neurodegenerative-diseases/ (accessed on 15 April 2022).
- ClinicalTrials.gov. A Study of BIIB067 When Initiated in Clinically Presymptomatic Adults With a Confirmed Superoxide Dismutase 1 Mutation (ATLAS). Available online: https://clinicaltrials.gov/ct2/show/NCT04856982 (accessed on 15 April 2022).
- Benatar, M.; Wuu, J.; Andersen, P.M.; Lombardi, V.; Malaspina, A. Neurofilament light: A candidate biomarker of presymptomatic amyotrophic lateral sclerosis and phenoconversion. Ann. Neurol. 2018, 84, 130–139. [Google Scholar] [CrossRef]
- Aoki, Y.; Wood, M.J.A. Emerging Oligonucleotide Therapeutics for Rare Neuromuscular Diseases. J. Neuromuscul. Dis. 2021, 8, 869–884. [Google Scholar] [CrossRef]
- Talbot, K.; Wood, M.J.A. Wrangling RNA: Antisense oligonucleotides for neurological disorders. Sci. Transl. Med. 2019, 11, eaay2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conte, A.; Lattante, S.; Zollino, M.; Marangi, G.; Luigetti, M.; Del Grande, A.; Servidei, S.; Trombetta, F.; Sabatelli, M. P525L FUS mutation is consistently associated with a severe form of juvenile amyotrophic lateral sclerosis. Neuromuscul. Disord. NMD 2012, 22, 73–75. [Google Scholar] [CrossRef] [PubMed]
- Neurology, P. Phase 3 Trial of Jacifusen for Amyloid Lateral Sclerosis With Fused-in-Sarcoma Mutation. Available online: https://practicalneurology.com/news/phase-3-trial-of-jacifusen-for-amyloid-lateral-sclerosis-with-fused-in-sarcoma-mutation#:~:text=04.07.21-,Phase%203%20Trial%20of%20Jacifusen%20for%20Amyloid,With%20Fused%2Din%2DSarcoma%20Mutation&text=A%20phase%203%20clinical%20trial,%2Dsarcoma%20gene%20(FUS) (accessed on 19 April 2022).
- ClinicalTrials.gov. A Study to Assess the Safety, Tolerability, and Pharmacokinetics of BIIB105 in Participants With Amyotrophic Lateral Sclerosis With or Without Poly-cytosine-adenine-guanine (CAG) Expansion in the Ataxin-2 Gene. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04494256?term=NCT04494256&type=Intr&cond=ALS&phase=012&draw=2&rank=1 (accessed on 19 April 2022).
- Elden, A.C.; Kim, H.J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D. Failure of genetic therapies for Huntington’s devastates community. Nature 2021, 593, 180. [Google Scholar] [CrossRef]
- Mizielinska, S.; Isaacs, A.M. C9orf72 amyotrophic lateral sclerosis and frontotemporal dementia: Gain or loss of function? Curr. Opin. Neurol. 2014, 27, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Belzil, V.V.; Bauer, P.O.; Prudencio, M.; Gendron, T.F.; Stetler, C.T.; Yan, I.K.; Pregent, L.; Daughrity, L.; Baker, M.C.; Rademakers, R.; et al. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 2013, 126, 895–905. [Google Scholar] [CrossRef]
- Pickar-Oliver, A.; Gersbach, C.A. The next generation of CRISPR-Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 2019, 20, 490–507. [Google Scholar] [CrossRef]
- Gugliandolo, A.; Bramanti, P.; Mazzon, E. Mesenchymal Stem Cells: A Potential Therapeutic Approach for Amyotrophic Lateral Sclerosis? Stem Cells Int. 2019, 2019, 3675627. [Google Scholar] [CrossRef]
- Zhao, C.P.; Zhang, C.; Zhou, S.N.; Xie, Y.M.; Wang, Y.H.; Huang, H.; Shang, Y.C.; Li, W.Y.; Zhou, C.; Yu, M.J.; et al. Human mesenchymal stromal cells ameliorate the phenotype of SOD1-G93A ALS mice. Cytotherapy 2007, 9, 414–426. [Google Scholar] [CrossRef]
- Vercelli, A.; Mereuta, O.M.; Garbossa, D.; Muraca, G.; Mareschi, K.; Rustichelli, D.; Ferrero, I.; Mazzini, L.; Madon, E.; Fagioli, F. Human mesenchymal stem cell transplantation extends survival, improves motor performance and decreases neuroinflammation in mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2008, 31, 395–405. [Google Scholar] [CrossRef]
- Uccelli, A.; Milanese, M.; Principato, M.C.; Morando, S.; Bonifacino, T.; Vergani, L.; Giunti, D.; Voci, A.; Carminati, E.; Giribaldi, F.; et al. Intravenous mesenchymal stem cells improve survival and motor function in experimental amyotrophic lateral sclerosis. Mol. Med. 2012, 18, 794–804. [Google Scholar] [CrossRef]
- Boido, M.; Piras, A.; Valsecchi, V.; Spigolon, G.; Mareschi, K.; Ferrero, I.; Vizzini, A.; Temi, S.; Mazzini, L.; Fagioli, F.; et al. Human mesenchymal stromal cell transplantation modulates neuroinflammatory milieu in a mouse model of amyotrophic lateral sclerosis. Cytotherapy 2014, 16, 1059–1072. [Google Scholar] [CrossRef]
- Forostyak, S.; Homola, A.; Turnovcova, K.; Svitil, P.; Jendelova, P.; Sykova, E. Intrathecal delivery of mesenchymal stromal cells protects the structure of altered perineuronal nets in SOD1 rats and amends the course of ALS. Stem Cells 2014, 32, 3163–3172. [Google Scholar] [CrossRef] [PubMed]
- Marconi, S.; Bonaconsa, M.; Scambi, I.; Squintani, G.M.; Rui, W.; Turano, E.; Ungaro, D.; D’Agostino, S.; Barbieri, F.; Angiari, S.; et al. Systemic treatment with adipose-derived mesenchymal stem cells ameliorates clinical and pathological features in the amyotrophic lateral sclerosis murine model. Neuroscience 2013, 248, 333–343. [Google Scholar] [CrossRef]
- Lewis, C.M.; Suzuki, M. Therapeutic applications of mesenchymal stem cells for amyotrophic lateral sclerosis. Stem Cell Res. Ther. 2014, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Mazzini, L.; Ferrero, I.; Luparello, V.; Rustichelli, D.; Gunetti, M.; Mareschi, K.; Testa, L.; Stecco, A.; Tarletti, R.; Miglioretti, M.; et al. Mesenchymal stem cell transplantation in amyotrophic lateral sclerosis: A Phase I clinical trial. Exp. Neurol. 2010, 223, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Mazzini, L.; Gelati, M.; Profico, D.C.; Sorarù, G.; Ferrari, D.; Copetti, M.; Muzi, G.; Ricciolini, C.; Carletti, S.; Giorgi, C.; et al. Results from Phase I Clinical Trial with Intraspinal Injection of Neural Stem Cells in Amyotrophic Lateral Sclerosis: A Long-Term Outcome. Stem Cells Transl. Med. 2019, 8, 887–897. [Google Scholar] [CrossRef]
- Glass, J.D.; Boulis, N.M.; Johe, K.; Rutkove, S.B.; Federici, T.; Polak, M.; Kelly, C.; Feldman, E.L. Lumbar intraspinal injection of neural stem cells in patients with amyotrophic lateral sclerosis: Results of a phase I trial in 12 patients. Stem Cells 2012, 30, 1144–1151. [Google Scholar] [CrossRef]
- Cudkowicz, M.E.; Lindborg, S.R.; Goyal, N.A.; Miller, R.G.; Burford, M.J.; Berry, J.D.; Nicholson, K.A.; Mozaffar, T.; Katz, J.S.; Jenkins, L.J.; et al. A randomized placebo-controlled phase 3 study of mesenchymal stem cells induced to secrete high levels of neurotrophic factors in amyotrophic lateral sclerosis. Muscle Nerve 2022, 65, 291–302. [Google Scholar] [CrossRef]
- Nabavi, S.M.; Arab, L.; Jarooghi, N.; Bolurieh, T.; Abbasi, F.; Mardpour, S.; Azimyian, V.; Moeininia, F.; Maroufizadeh, S.; Sanjari, L.; et al. Safety, Feasibility of Intravenous and Intrathecal Injection of Autologous Bone Marrow Derived Mesenchymal Stromal Cells in Patients with Amyotrophic Lateral Sclerosis: An Open Label Phase I Clinical Trial. Cell J. 2019, 20, 592–598. [Google Scholar] [CrossRef]
- Oh, K.W.; Noh, M.Y.; Kwon, M.S.; Kim, H.Y.; Oh, S.I.; Park, J.; Kim, H.J.; Ki, C.S.; Kim, S.H. Repeated Intrathecal Mesenchymal Stem Cells for Amyotrophic Lateral Sclerosis. Ann. Neurol. 2018, 84, 361–373. [Google Scholar] [CrossRef]
- Staff, N.P.; Madigan, N.N.; Morris, J.; Jentoft, M.; Sorenson, E.J.; Butler, G.; Gastineau, D.; Dietz, A.; Windebank, A.J. Safety of intrathecal autologous adipose-derived mesenchymal stromal cells in patients with ALS. Neurology 2016, 87, 2230–2234. [Google Scholar] [CrossRef] [PubMed]
- Petrou, P.; Gothelf, Y.; Argov, Z.; Gotkine, M.; Levy, Y.S.; Kassis, I.; Vaknin-Dembinsky, A.; Ben-Hur, T.; Offen, D.; Abramsky, O.; et al. Safety and Clinical Effects of Mesenchymal Stem Cells Secreting Neurotrophic Factor Transplantation in Patients With Amyotrophic Lateral Sclerosis: Results of Phase 1/2 and 2a Clinical Trials. JAMA Neurol. 2016, 73, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Barczewska, M.; Grudniak, M.; Maksymowicz, S.; Siwek, T.; Ołdak, T.; Jezierska-Woźniak, K.; Gładysz, D.; Maksymowicz, W. Safety of intrathecal injection of Wharton’s jelly-derived mesenchymal stem cells in amyotrophic lateral sclerosis therapy. Neural Regen. Res. 2019, 14, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Siwek, T.; Maksymowicz, W.; Barczewska, M.; Grabarczyk, L.; Sowa, M.J.J.N.N. Mesenchymal stem cell (MSC) transplantation in patients with amyotrophic lateral sclerosis (ALS): Is there a “responder population”. J. Neurol. Neurosci. 2018, 9, 260. [Google Scholar] [CrossRef]
- Scott, S.; Kranz, J.E.; Cole, J.; Lincecum, J.M.; Thompson, K.; Kelly, N.; Bostrom, A.; Theodoss, J.; Al-Nakhala, B.M.; Vieira, F.G.; et al. Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph. Lateral Scler. Off. Publ. World Fed. Neurol. Res. Group Mot. Neuron Dis. 2008, 9, 4–15. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Cleveland, D.W. Rethinking ALS: The FUS about TDP-43. Cell 2009, 136, 1001–1004. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Ripps, M.E.; Huntley, G.W.; Hof, P.R.; Morrison, J.H.; Gordon, J.W. Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 1995, 92, 689–693. [Google Scholar] [CrossRef]
- Chiang, P.M.; Ling, J.; Jeong, Y.H.; Price, D.L.; Aja, S.M.; Wong, P.C. Deletion of TDP-43 down-regulates Tbc1d1, a gene linked to obesity, and alters body fat metabolism. Proc. Natl. Acad. Sci. USA 2010, 107, 16320–16324. [Google Scholar] [CrossRef]
- Wegorzewska, I.; Bell, S.; Cairns, N.J.; Miller, T.M.; Baloh, R.H. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 18809–18814. [Google Scholar] [CrossRef] [Green Version]
- Stribl, C.; Samara, A.; Trümbach, D.; Peis, R.; Neumann, M.; Fuchs, H.; Gailus-Durner, V.; Hrabě de Angelis, M.; Rathkolb, B.; Wolf, E.; et al. Mitochondrial dysfunction and decrease in body weight of a transgenic knock-in mouse model for TDP-43. J. Biol. Chem. 2014, 289, 10769–10784. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, M.; Sok, J.; Webb, L.; Baechtold, H.; Urano, F.; Yin, Y.; Chung, P.; de Rooij, D.G.; Akhmedov, A.; Ashley, T.; et al. Male sterility and enhanced radiation sensitivity in TLS(-/-) mice. EMBO J. 2000, 19, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Hicks, G.G.; Singh, N.; Nashabi, A.; Mai, S.; Bozek, G.; Klewes, L.; Arapovic, D.; White, E.K.; Koury, M.J.; Oltz, E.M.; et al. Fus deficiency in mice results in defective B-lymphocyte development and activation, high levels of chromosomal instability and perinatal death. Nat. Genet. 2000, 24, 175–179. [Google Scholar] [CrossRef]
- Mitchell, J.C.; McGoldrick, P.; Vance, C.; Hortobagyi, T.; Sreedharan, J.; Rogelj, B.; Tudor, E.L.; Smith, B.N.; Klasen, C.; Miller, C.C.; et al. Overexpression of human wild-type FUS causes progressive motor neuron degeneration in an age- and dose-dependent fashion. Acta Neuropathol. 2013, 125, 273–288. [Google Scholar] [CrossRef]
- Scekic-Zahirovic, J.; Sendscheid, O.; El Oussini, H.; Jambeau, M.; Sun, Y.; Mersmann, S.; Wagner, M.; Dieterlé, S.; Sinniger, J.; Dirrig-Grosch, S.; et al. Toxic gain of function from mutant FUS protein is crucial to trigger cell autonomous motor neuron loss. EMBO J. 2016, 35, 1077–1097. [Google Scholar] [CrossRef] [PubMed]
- Koppers, M.; Blokhuis, A.M.; Westeneng, H.J.; Terpstra, M.L.; Zundel, C.A.; Vieira de Sá, R.; Schellevis, R.D.; Waite, A.J.; Blake, D.J.; Veldink, J.H.; et al. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann. Neurol. 2015, 78, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Mizielinska, S.; Grönke, S.; Niccoli, T.; Ridler, C.E.; Clayton, E.L.; Devoy, A.; Moens, T.; Norona, F.E.; Woollacott, I.O.C.; Pietrzyk, J.; et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 2014, 345, 1192–1194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef]
- Trudler, D.; Ghatak, S.; Lipton, S.A. Emerging hiPSC Models for Drug Discovery in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 8196. [Google Scholar] [CrossRef]
- Egawa, N.; Kitaoka, S.; Tsukita, K.; Naitoh, M.; Takahashi, K.; Yamamoto, T.; Adachi, F.; Kondo, T.; Okita, K.; Asaka, I.; et al. Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci. Transl. Med. 2012, 4, 145ra104. [Google Scholar] [CrossRef]
- Okano, H.; Yasuda, D.; Fujimori, K.; Morimoto, S.; Takahashi, S. Ropinirole, a New ALS Drug Candidate Developed Using iPSCs. Trends Pharmacol. Sci. 2020, 41, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, G.; Iemmolo, R.; Attaguile, G.A.; La Cognata, V.; Pistone, B.S.; Raudino, G.; D’Agata, V.; Cantarella, G.; Barcellona, M.L.; Cavallaro, S. iPSCs: A Preclinical Drug Research Tool for Neurological Disorders. Int. J. Mol. Sci. 2021, 22, 4596. [Google Scholar] [CrossRef] [PubMed]
- Goyal, N.A.; Berry, J.D.; Windebank, A.; Staff, N.P.; Maragakis, N.J.; van den Berg, L.H.; Genge, A.; Miller, R.; Baloh, R.H.; Kern, R.; et al. Addressing heterogeneity in amyotrophic lateral sclerosis CLINICAL TRIALS. Muscle Nerve 2020, 62, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Andrews, J.A.; Bruijn, L.I.; Shefner, J.M. ALS drug development guidances and trial guidelines: Consensus and opportunities for alignment. Neurology 2019, 93, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Westeneng, H.J.; Debray, T.P.A.; Visser, A.E.; van Eijk, R.P.A.; Rooney, J.P.K.; Calvo, A.; Martin, S.; McDermott, C.J.; Thompson, A.G.; Pinto, S.; et al. Prognosis for patients with amyotrophic lateral sclerosis: Development and validation of a personalised prediction model. Lancet. Neurol. 2018, 17, 423–433. [Google Scholar] [CrossRef]
- Taylor, A.A.; Fournier, C.; Polak, M.; Wang, L.; Zach, N.; Keymer, M.; Glass, J.D.; Ennist, D.L. Predicting disease progression in amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2016, 3, 866–875. [Google Scholar] [CrossRef]
- Taga, A.; Maragakis, N.J. Current and emerging ALS biomarkers: Utility and potential in clinical trials. Expert Rev. Neurother. 2018, 18, 871–886. [Google Scholar] [CrossRef]
- Steinacker, P.; Huss, A.; Mayer, B.; Grehl, T.; Grosskreutz, J.; Borck, G.; Kuhle, J.; Lulé, D.; Meyer, T.; Oeckl, P.; et al. Diagnostic and prognostic significance of neurofilament light chain NF-L, but not progranulin and S100B, in the course of amyotrophic lateral sclerosis: Data from the German MND-net. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 112–119. [Google Scholar] [CrossRef]
- Bromberg, M.B. MUNIX and MUNE in ALS. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2013, 124, 433–434. [Google Scholar] [CrossRef]
- Vucic, S.; Nicholson, G.A.; Kiernan, M.C. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain A J. Neurol. 2008, 131, 1540–1550. [Google Scholar] [CrossRef] [Green Version]
- Foerster, B.R.; Welsh, R.C.; Feldman, E.L. 25 years of neuroimaging in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2013, 9, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gancedo, L.; Kelly, M.L.; Lavrov, A.; Parr, J.; Hart, R.; Marsden, R.; Turner, M.R.; Talbot, K.; Chiwera, T.; Shaw, C.E.; et al. Objectively Monitoring Amyotrophic Lateral Sclerosis Patient Symptoms During Clinical Trials With Sensors: Observational Study. JMIR Mhealth Uhealth 2019, 7, e13433. [Google Scholar] [CrossRef] [PubMed]
- Barkhof, F. MRI in multiple sclerosis: Correlation with expanded disability status scale (EDSS). Mult. Scler. J. 1999, 5, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Brück, W.; Bitsch, A.; Kolenda, H.; Brück, Y.; Stiefel, M.; Lassmann, H. Inflammatory central nervous system demyelination: Correlation of magnetic resonance imaging findings with lesion pathology. Ann. Neurol. 1997, 42, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Katz, D.; Taubenberger, J.K.; Cannella, B.; McFarlin, D.E.; Raine, C.S.; McFarland, H.F. Correlation between magnetic resonance imaging findings and lesion development in chronic, active multiple sclerosis. Ann. Neurol. 1993, 34, 661–669. [Google Scholar] [CrossRef]
- van Dyck, C.H. Anti-Amyloid-β Monoclonal Antibodies for Alzheimer’s Disease: Pitfalls and Promise. Biol. Psychiatry 2018, 83, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Massachusetts General Hospital. HEALEY ALS Platform Trial. Available online: https://www.massgeneral.org/neurology/als/research/platform-trial (accessed on 29 April 2022).



{kind=link}
{kind=link}
| Agent | Targeted Mechanism | Mechanism | Results | Phase | Ref. |
|---|---|---|---|---|---|
| Sodium Phenylbutyrate-Taurursodiol | endoplasmic reticulum stress, and mitochondrial dysfunction | Sodium phenylbutyrate is a histone deacetylase inhibitor that has been shown to upregulate heat shock proteins and act as a small molecule chaperone, alleviating endoplasmic reticulum stress toxicity [17,18]. Taurursodiol recovers mitochondrial bioenergetic deficits through multiple mechanisms, including preventing the translocation of Bax protein into the mitochondrial membrane, thereby decreasing mitochondrial permeability and increasing the cell’s apoptotic threshold [19] | Less functional deterioration measured by the ALSFRS-R score over a 24-week period. Secondary outcomes, including decreases in isometric muscle strength and vital capacity, did not differ significantly between groups | II | [20] |
| Colchicine | Protein aggregates, autophagy, and neuroinflammation | Colchicine could upregulate proteins involved in autophagy, including the TFEB, the TFEB-regulated adaptor protein SQSTM1/p62 and the autophagy player microtubule-associated protein 1A/1B-light chain 3 (LC3). | Ongoing | II | [21] |
| Rapamycin | Autophagy and neuroinflammation | Rapamycin is based on the inhibition of mTORC1. mTORC1 targets regulatory proteins in cell signalling and regulates autophagy by inhibiting the unc-51-like kinase 1 complex. | Ongoing | II | [22] |
| BIIB100 (KPT-350) | Nucleocytoplasmic transport dysfunction | Selective inhibitor of nuclear export that inhibits exportin 1 (XPO1; CRM1). | Ongoing | I | |
| Deferiprone | Iron accumulation | Iron Chelation | Ongoing | II | [23] |
| TIRASEMTIV | Muscle contractility | A FSTA that selectively activates the fast skeletal muscle troponin complex by increasing its sensitivity to calcium | In a phase IIb clinical trial, SVC and muscle strength were found to decline significantly more slowly in tirasemtiv-treated participants. But no significant difference was found in the decline in functional disability as measured by the ALSFRS-R. However, no significant difference in disease progression was demonstrated in the phase III clinical trial. | II/III | [24,25] |
| Interleukine 2 | Neuroinflammation | Immunomodulatory strategy by promoting Treg expansion, which attenuates neuroinflammation. | A phase IIa study showed that low dose IL-2 is well tolerated and immunologically effective in subjects with ALS [26] | III | [26] |
| Masitinib | Neuroinflammation | Tyrosine kinase inhibitor targets microglia and mast cells through inhibiting a limited number of kinases. Masitinib blocks microglia proliferation and activation, and mast cell-mediated degranulation, the release of cytotoxic substances that might further damage the motor nerves. | A randomised, placebo-controlled phase III trial has previously shown that oral masitinib (4.5 mg/kg/day) slows the rate of functional decline with acceptable safety in ALS patients with an ALSFRS-R progression rate of <1.1 points/month | III | [27] |
| Ibudilast (MN-166) | Neuroinflammation | Inhibitor of macrophage migration inhibitory factor and phosphodiesterases 3,4,10 and 11 [28,29]. Ibudilast attenuates CNS microglial activation and secretion of pro-inflammatory cytokines. | Ongoing | II/III | [29,30] |
| Fasudil | Neuroinflammation | Rho kinase inhibitor | Ongoing | II | [31] |
| Ravulizumab | Neuroinflammation | Humanized monoclonal antibody to complement factor 5 which acts to block complement activation | The independent Data and Safety Monitoring Board monitoring committee recommended that the study be discontinued due to lack of efficacy. No new safety findings were observed. | III | [32] |
| Zilucoplan | Neuroinflammation | A small molecule that works aa s C5 complement inhibitor | The The independent Data and Safety Monitoring Board recommended stopping the zilucoplan regimen because the likelihood of meaningfully slowing disease progression was considered low. | III | [33] |
| Anakinra | Neuroinflammation | The monoclonal antibody that works as a IL–1 receptor antagonist | Ongoing | II | |
| Tocilizumab | Neuroinflammation | The monoclonal antibody that works as a IL–1 receptor antagonist | Tocilizumab is safe and tolerable and reduces C-reactive protein concentrations in the plasma and cerebrospinal fluid of ALS patients | II | [34] |
| Tofersen (BIIB067) | Gain of function SOD1 | It is an antisense oligonucleotide (ASO) targeting SOD1 | In the Phase III VALOR study, the primary endpoint as measured by the ALSFRS-R did not reach statistical significance; however, signs of reduced disease progression across multiple secondary and exploratory endpoints were observed | III | [35] |
| BIIB078 | Gain of function C9ORF72 | It is an antisense oligonucleotide (ASO) for C9ORF72-associated ALS | In a Phase I study, BIIB078 was generally well-tolerated. The adverse events were mostly mild to moderate in severity and occurred at a similar rate across BIIB078 and placebo groups. BIIB078 did not meet any secondary efficacy endpoints and it did not demonstrate clinical benefit. Therefore, the clinical program will be discontinued | I | [36] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khamaysa, M.; Pradat, P.-F. Status of ALS Treatment, Insights into Therapeutic Challenges and Dilemmas. J. Pers. Med. 2022, 12, 1601. https://doi.org/10.3390/jpm12101601
Khamaysa M, Pradat P-F. Status of ALS Treatment, Insights into Therapeutic Challenges and Dilemmas. Journal of Personalized Medicine. 2022; 12(10):1601. https://doi.org/10.3390/jpm12101601
Chicago/Turabian StyleKhamaysa, Mohammed, and Pierre-François Pradat. 2022. "Status of ALS Treatment, Insights into Therapeutic Challenges and Dilemmas" Journal of Personalized Medicine 12, no. 10: 1601. https://doi.org/10.3390/jpm12101601