
Molecular Classification Models for Triple Negative Breast Cancer Subtype Using Machine Learning

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
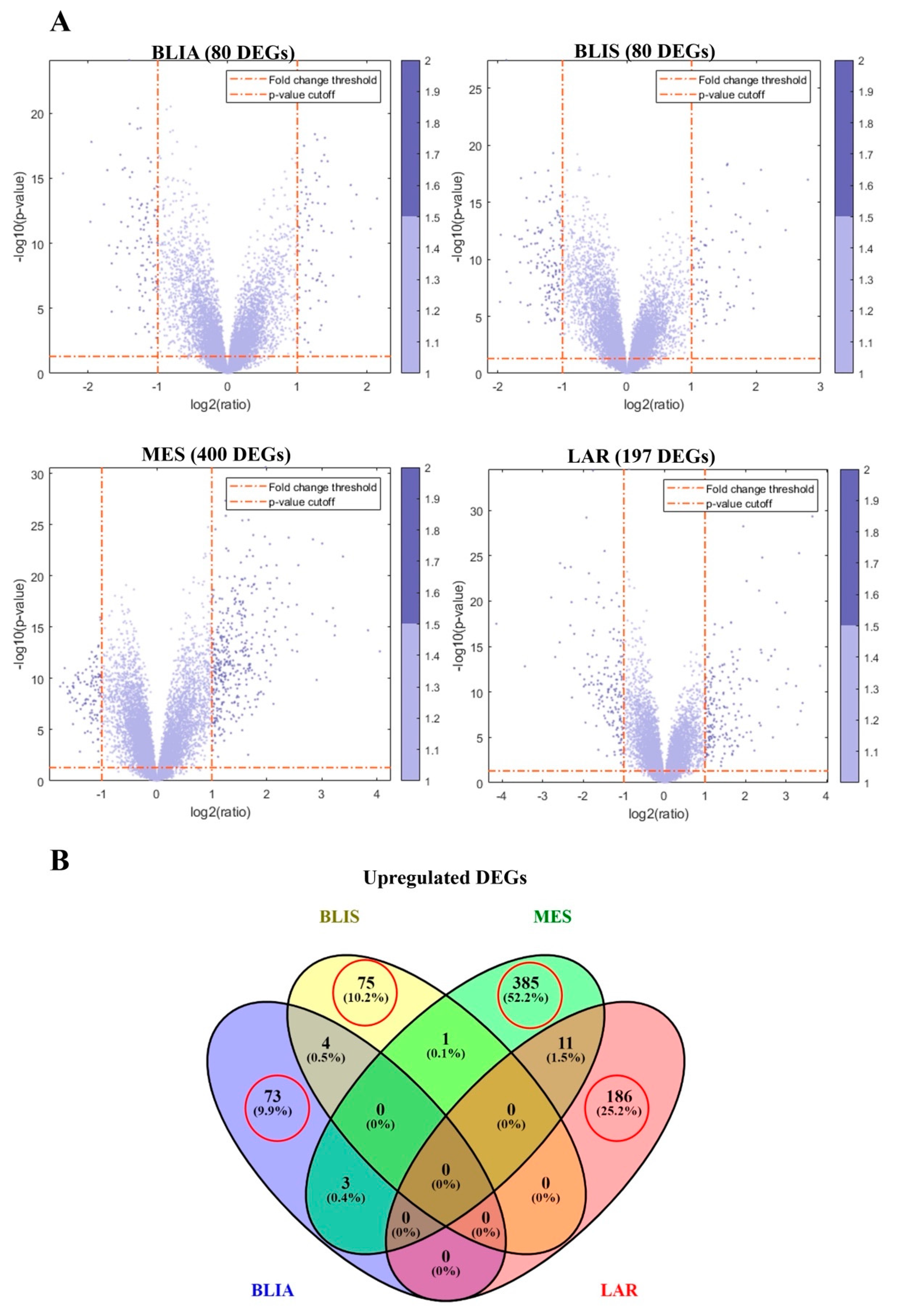
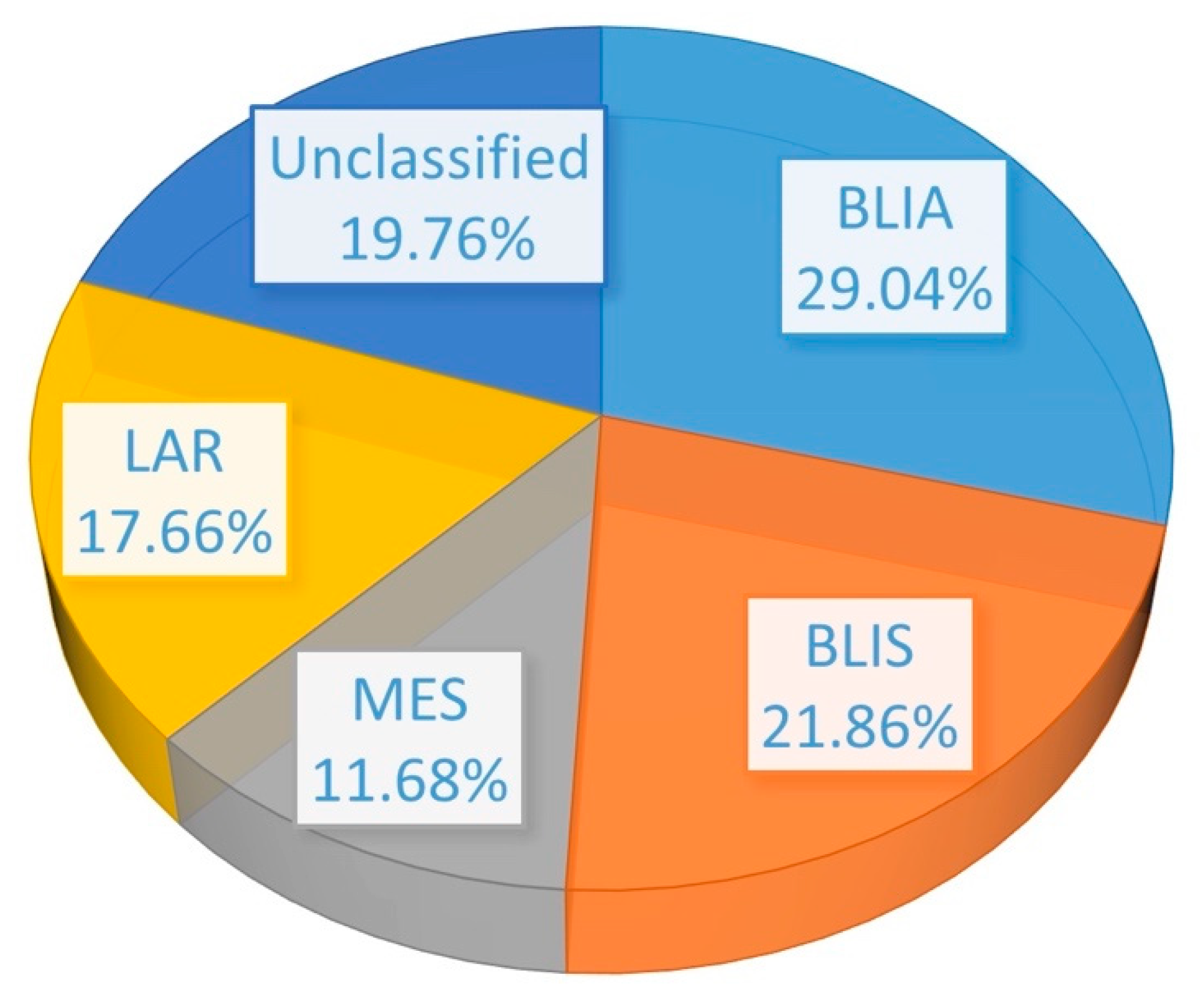
2.1. Identification of DEGs and Feature Selection
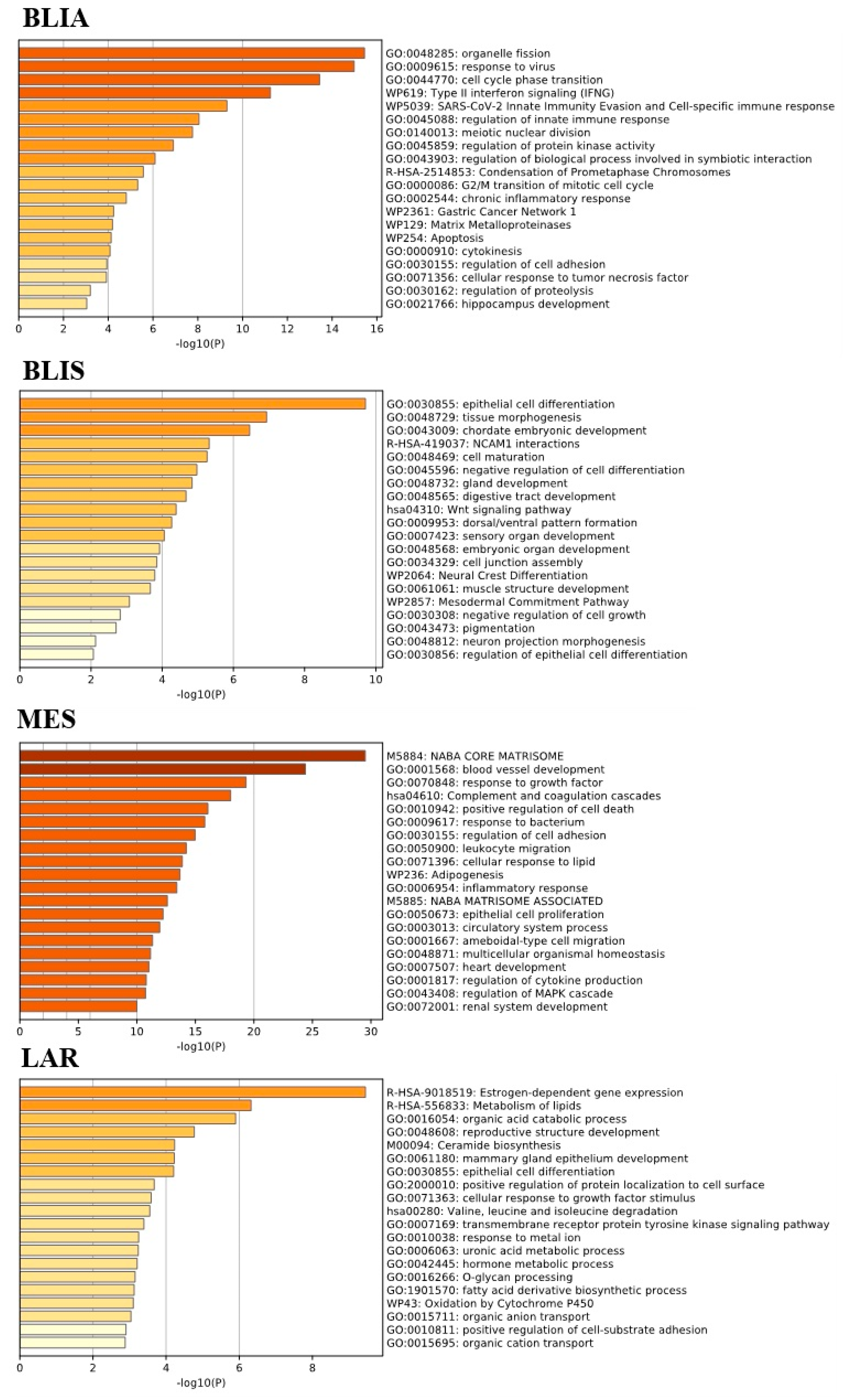
2.2. GO Term and KEGG Pathway Enrichment Analysis of Unique Upregulated DEGs in Each TNBC Subtype
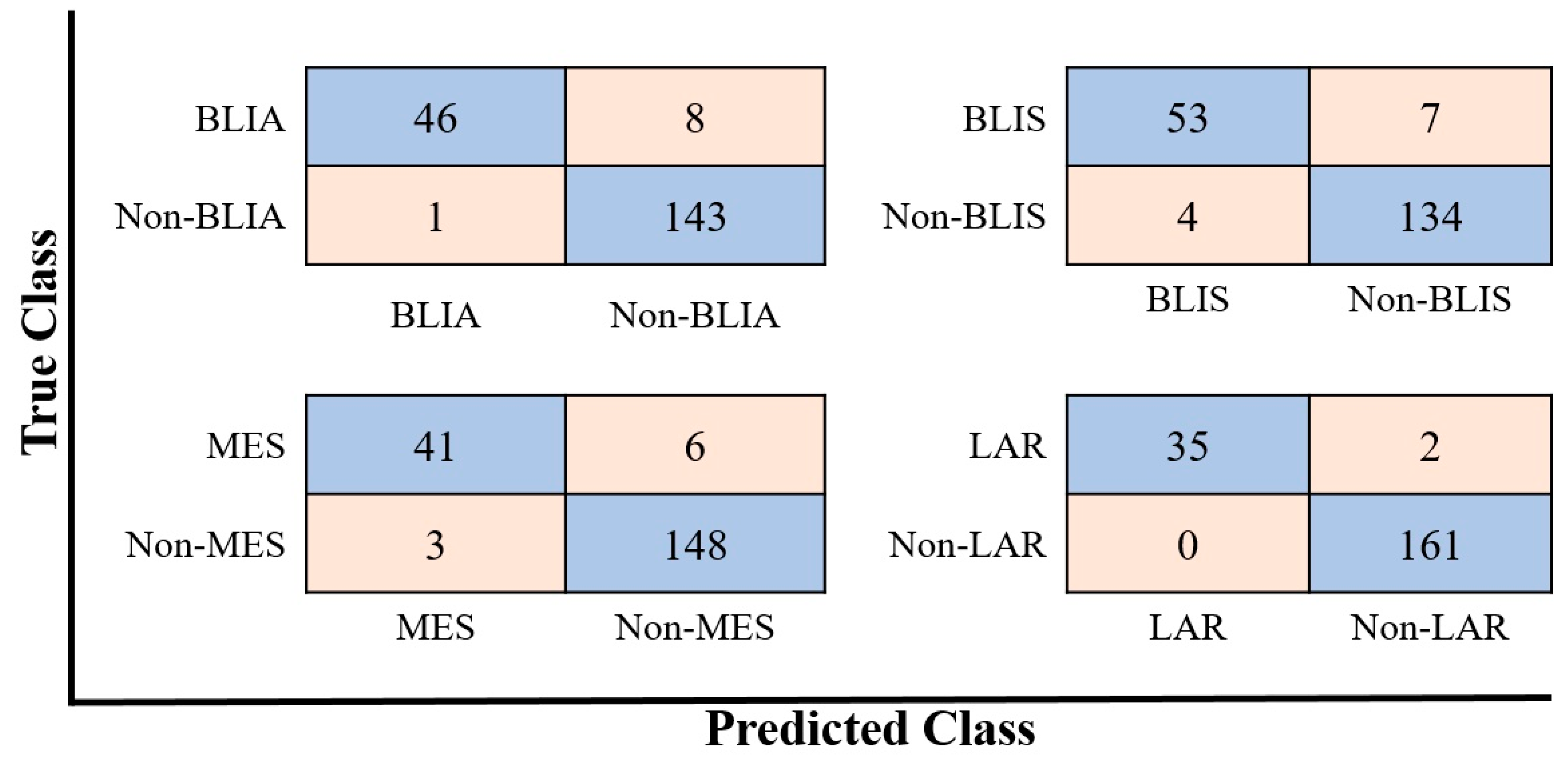
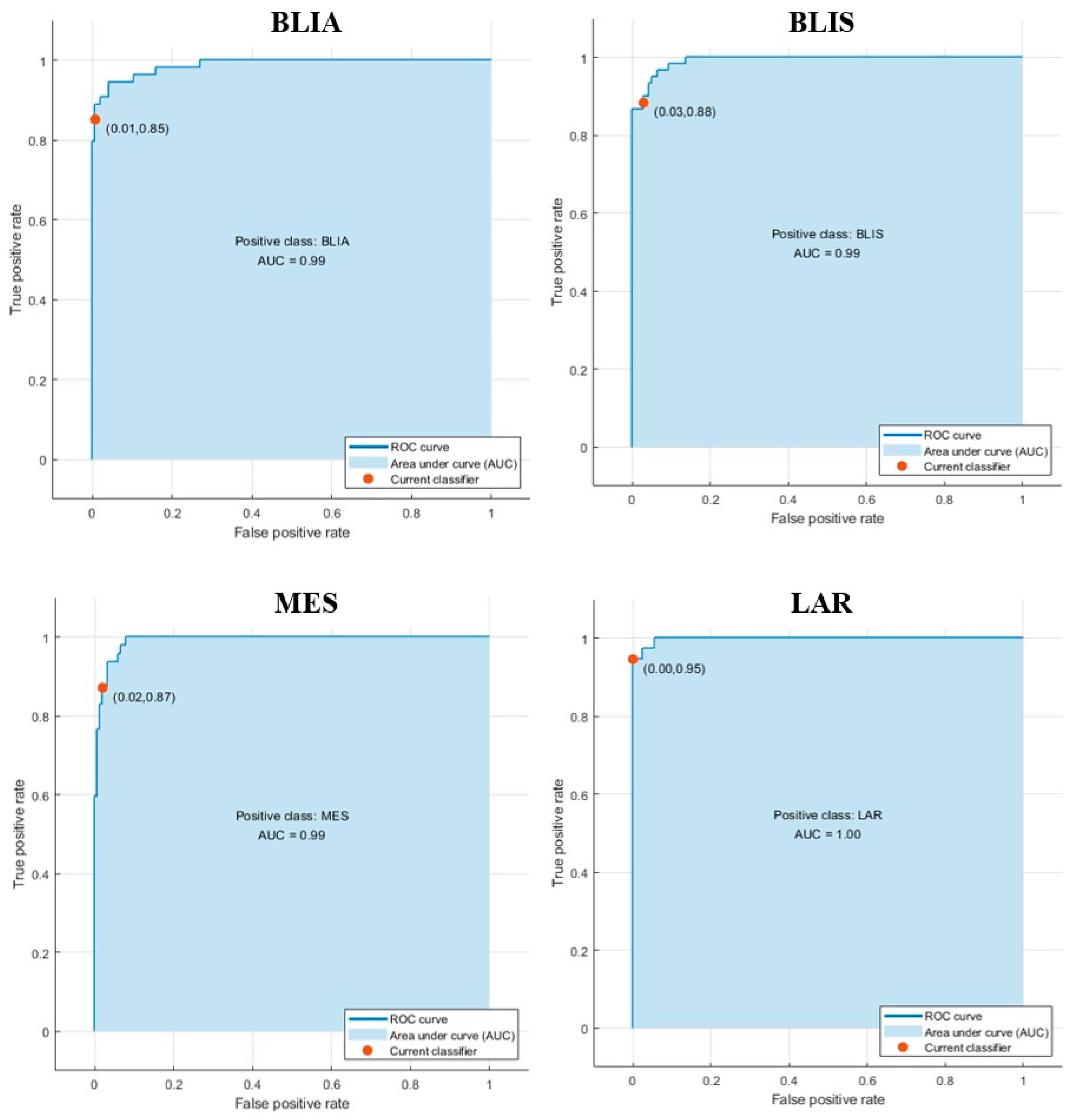
2.3. Modeling Prediction and Performance Evaluation
2.4. Testing on Independent Cohorts Demonstrated the Generalizability of the Classification Model
3. Discussion
4. Materials and Methods
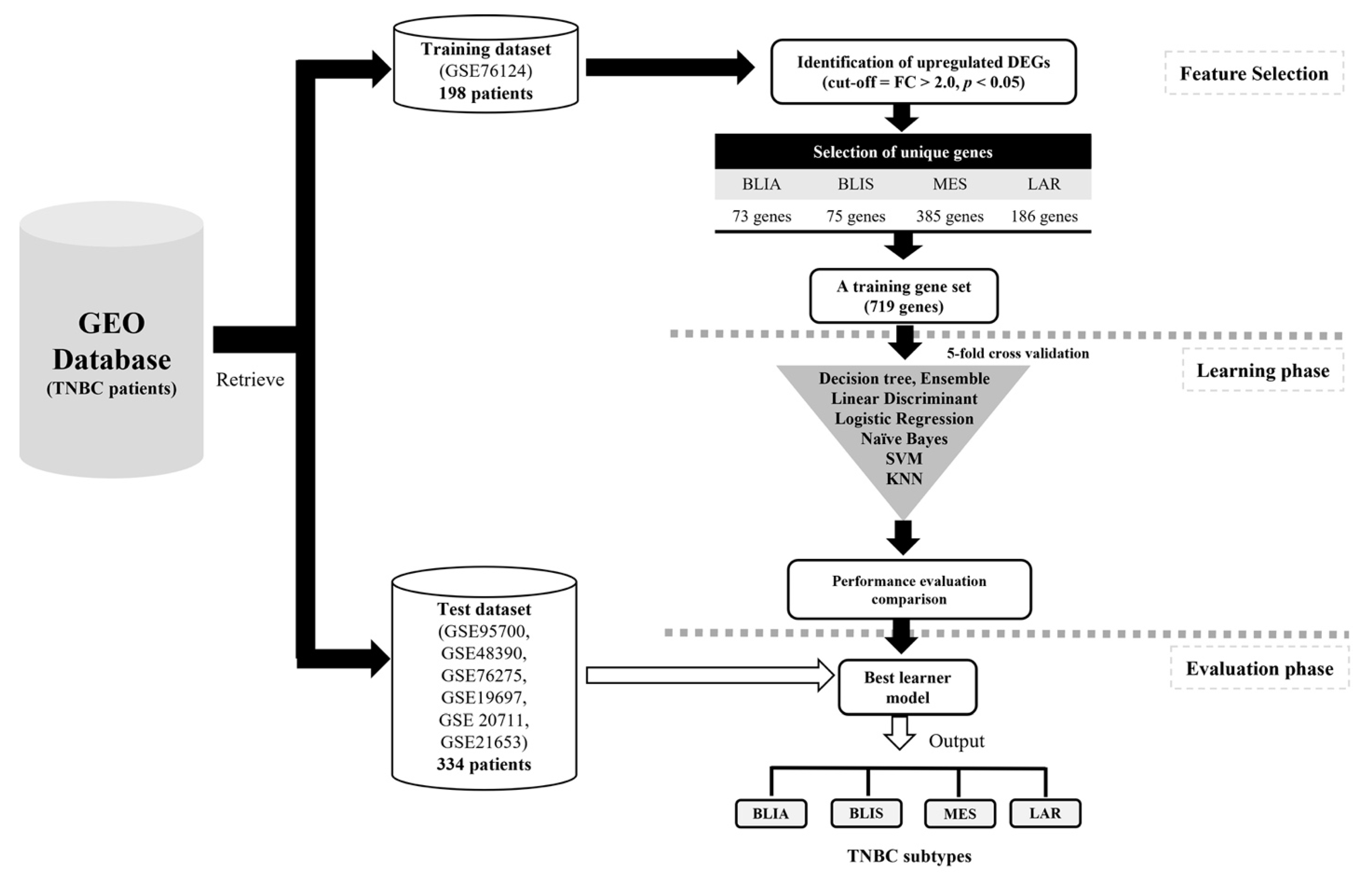
4.1. Data Sources and Preprocessing
4.2. Identification of DEGs and Feature Selection
4.3. Functional and Pathway Enrichment Analysis
4.4. Model Construction
Sensitivity (Recall) = TP/(TP + FN)
Specificity = TN/(TN + FP)
Precision (positive predictive value (PPV)) = TP/(TP + FP)
Negative predictive value (NPV)) = TN/(TN + FN)
F1 score = 2(Precision* Recall)/(Precision + Recall)
where, TP = true positive, TN = true negative, FN = false negative,
FP = false positive.
4.5. Modeling Prediction and Performance Evaluation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yao, H.; He, G.; Yan, S.; Chen, C.; Song, L.; Rosol, T.J.; Deng, X. Triple-negative breast cancer: Is there a treatment on the horizon? Oncotarget 2017, 8, 1913–1924. [Google Scholar] [CrossRef] [Green Version]
- Schmadeka, R.; Harmon, B.E.; Singh, M. Triple-Negative Breast Carcinoma: Current and Emerging Concepts. Am. J. Clin. Pathol. 2014, 141, 462–477. [Google Scholar] [CrossRef] [Green Version]
- Millis, S.Z.; Gatalica, Z.; Winkler, J.; Vranic, S.; Kimbrough, J.; Reddy, S.; O’Shaughnessy, J.A. Predictive Biomarker Profiling of >6000 Breast Cancer Patients Shows Heterogeneity in TNBC, with Treatment Implications. Clin. Breast Cancer 2015, 15, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Yoshimaru, T.; Matsuo, T.; Kiyotani, K.; Miyoshi, Y.; Tanahashi, T.; Rokutan, K.; Yamaguchi, R.; Saito, A.; Imoto, S.; et al. Molecular features of triple negative breast cancer cells by genome-wide gene expression profiling analysis. Int. J. Oncol. 2013, 42, 478–506. [Google Scholar] [CrossRef] [Green Version]
- Al-Ejeh, F.; Simpson, P.T.; Sanus, J.M.; Klein, K.; Kalimutho, M.; Shi, W.; Miranda, M.; Kutasovic, J.; Raghavendra, A.; Madore, J.; et al. Meta-analysis of the global gene expression profile of triple-negative breast cancer identifies genes for the prognostication and treatment of aggressive breast cancer. Oncogenesis 2014, 3, e100. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.W.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef] [Green Version]
- Masuda, H.; Baggerly, K.A.; Wang, Y.; Zhang, Y.; Gonzalez-Angulo, A.M.; Meric-Bernstam, F.; Valero, V.; Lehmann, B.D.; Pietenpol, J.A.; Hortobagyi, G.N.; et al. Differential response to neoadjuvant chemotherapy among 7 triple-negative breast cancer molecular subtypes. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 5533–5540. [Google Scholar] [CrossRef] [Green Version]
- Hubalek, M.; Czech, T.; Müller, H. Biological Subtypes of Triple-Negative Breast Cancer. Breast Care 2017, 12, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Zhang, L.; Wang, B.; Li, F.; Yang, J. A fast gene selection method for multi-cancer classification using multiple support vector data description. J. Biomed. Inform. 2015, 53, 381–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.H.; Iqbal, M.J.; Ahmad, I.; Khan, S.; Rodrigues, J.J.P.C. Optimized gene selection and classification of cancer from microarray gene expression data using deep learning. Neural Comput. Appl. 2020. [Google Scholar] [CrossRef]
- Thomas, M.M. Machine Learning; McGraw-Hill, Inc. Professional Book Group 11 West 19th Street New York, NY, United States: New York, NY, USA, 1997; ISBN 978-0-07-042807-2. [Google Scholar]
- Xu, Y.; Ju, L.; Tong, J.; Zhou, C.; Yang, J. Supervised Machine Learning Predictive Analytics For Triple-Negative Breast Cancer Death Outcomes. OncoTargets Ther. 2019, 12, 9059–9067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athreya, A.P.; Gaglio, A.J.; Cairns, J.; Kalari, K.R.; Weinshilboum, R.M.; Wang, L.; Kalbarczyk, Z.T.; Iyer, R.K. Machine Learning Helps Identify New Drug Mechanisms in Triple-Negative Breast Cancer. IEEE Trans. Nanobioscience 2018, 17, 251–259. [Google Scholar] [CrossRef]
- Kothari, C.; Osseni, M.A.; Agbo, L.; Ouellette, G.; Déraspe, M.; Laviolette, F.; Corbeil, J.; Lambert, J.-P.; Diorio, C.; Durocher, F. Machine learning analysis identifies genes differentiating triple negative breast cancers. Sci. Rep. 2020, 10, 10464. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets–update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- MATLAB (2020) Version 9.8 (R2020a); The MathWorks Inc.: Natick, MA, USA, 2020.
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [Green Version]
- Lin, N.U.; Claus, E.; Sohl, J.; Razzak, A.R.; Arnaout, A.; Winer, E.P. Sites of distant recurrence and clinical outcomes in patients with metastatic triple-negative breast cancer: High incidence of central nervous system metastases. Cancer 2008, 113, 2638–2645. [Google Scholar] [CrossRef] [Green Version]
- Abramson, V.G.; Lehmann, B.D.; Ballinger, T.J.; Pietenpol, J.A. Subtyping of triple-negative breast cancer: Implications for therapy. Cancer 2015, 121, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.C.; Steele, L.; Warden, C.; Wilczynski, S.; Mortimer, J.; Yuan, Y.; Neuhausen, S.L. Molecular subtypes of triple-negative breast cancer in women of different race and ethnicity. Oncotarget 2019, 10, 198–208. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-R.; Jiang, Y.-Z.; Xu, X.-E.; Yu, K.-D.; Jin, X.; Hu, X.; Zuo, W.-J.; Hao, S.; Wu, J.; Liu, G.-Y.; et al. Comprehensive transcriptome analysis identifies novel molecular subtypes and subtype-specific RNAs of triple-negative breast cancer. Breast Cancer Res. 2016, 18, 33. [Google Scholar] [CrossRef] [Green Version]
- Santonja, A.; Sánchez-Muñoz, A.; Lluch, A.; Chica-Parrado, M.R.; Albanell, J.; Chacón, J.I.; Antolín, S.; Jerez, J.M.; de la Haba, J.; de Luque, V.; et al. Triple negative breast cancer subtypes and pathologic complete response rate to neoadjuvant chemotherapy. Oncotarget 2018, 9, 26406–26416. [Google Scholar] [CrossRef] [Green Version]
- Asri, H.; Mousannif, H.; Al Moatassime, H.; Noel, T. Using Machine Learning Algorithms for Breast Cancer Risk Prediction and Diagnosis. Procedia Comput. Sci. 2016, 83, 1064–1069. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Hicks, C. Breast Cancer Type Classification Using Machine Learning. J. Pers. Med. 2021, 11, 61. [Google Scholar] [CrossRef]
- Nindrea, R.D.; Aryandono, T.; Lazuardi, L.; Dwiprahasto, I. Diagnostic Accuracy of Different Machine Learning Algorithms for Breast Cancer Risk Calculation: A Meta-Analysis. Asian Pac. J. Cancer Prev. 2018, 19, 1747–1752. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Duan, J.-J.; Bian, X.-W.; Yu, S. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef] [PubMed]
- Welboren, W.-J.; Sweep, F.C.G.J.; Span, P.N.; Stunnenberg, H.G. Genomic actions of estrogen receptor alpha: What are the targets and how are they regulated? Endocr. -Relat. Cancer 2009, 16, 1073–1089. [Google Scholar] [CrossRef] [Green Version]
- Gerratana, L.; Basile, D.; Buono, G.; De Placido, S.; Giuliano, M.; Minichillo, S.; Coinu, A.; Martorana, F.; De Santo, I.; Del Mastro, L.; et al. Androgen receptor in triple negative breast cancer: A potential target for the targetless subtype. Cancer Treat. Rev. 2018, 68, 102–110. [Google Scholar] [CrossRef]
- Brumec, M.; Sobočan, M.; Takač, I.; Arko, D. Clinical Implications of Androgen-Positive Triple-Negative Breast Cancer. Cancers 2021, 13, 1642. [Google Scholar] [CrossRef]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. affy—Analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Ruepp, A.; Brauner, B.; Dunger-Kaltenbach, I.; Frishman, G.; Montrone, C.; Stransky, M.; Waegele, B.; Schmidt, T.; Doudieu, O.N.; Stümpflen, V.; et al. CORUM: The comprehensive resource of mammalian protein complexes. Nucleic Acids Res. 2008, 36, D646–D650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Powers, D. Ailab Evaluation: From precision, recall and F-measure to ROC, informedness, markedness & correlation. J. Mach. Learn. Technol. 2011, 2, 2229–3981. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
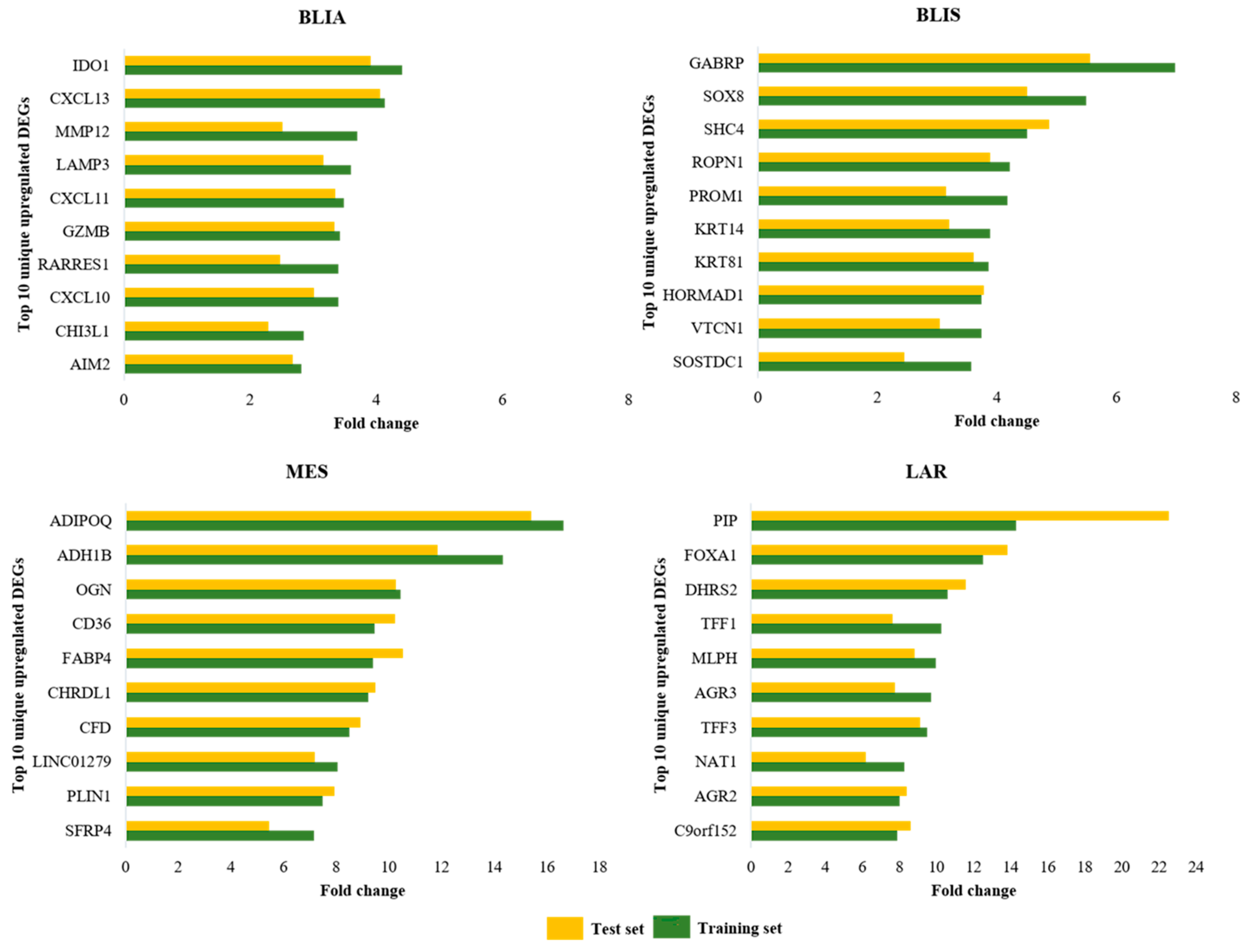
| BLIA | BLIS | MES | LAR | ||||
|---|---|---|---|---|---|---|---|
| DEGs | FC | DEGs | FC | DEGs | FC | DEGs | FC |
| IDO1 | 4.41 | GABRP | 6.97 | ADIPOQ | 16.63 | PIP | 14.29 |
| CXCL13 | 4.13 | SOX8 | 5.49 | ADH1B | 14.32 | FOXA1 | 12.52 |
| MMP12 | 3.69 | SHC4 | 4.51 | OGN | 10.44 | DHRS2 | 10.59 |
| LAMP3 | 3.60 | ROPN1 | 4.21 | CD36 | 9.46 | TFF1 | 10.27 |
| CXCL11 | 3.49 | PROM1 | 4.17 | FABP4 | 9.40 | MLPH | 9.97 |
| GZMB | 3.43 | KRT14 | 3.89 | CHRDL1 | 9.21 | AGR3 | 9.70 |
| RARRES1 | 3.40 | KRT81 | 3.86 | CFD | 8.50 | TFF3 | 9.48 |
| CXCL10 | 3.40 | HORMAD1 | 3.75 | LINC01279 | 8.05 | NAT1 | 8.27 |
| CHI3L1 | 2.85 | VTCN1 | 3.74 | PLIN1 | 7.49 | AGR2 | 8.01 |
| AIM2 | 2.81 | SOSTDC1 | 3.57 | SFRP4 | 7.15 | C9orf152 | 7.87 |
| NUF2 | 2.71 | PNMA8A | 3.50 | ACKR1 | 7.11 | SCUBE2 | 6.99 |
| TTK | 2.70 | IRX1 | 3.47 | IGF1 | 5.95 | GATA3 | 6.86 |
| CXCL9 | 2.66 | UGT8 | 3.38 | HBB | 5.86 | SIDT1 | 6.79 |
| APOBEC3B | 2.65 | KRT23 | 3.31 | EFEMP1 | 5.84 | REEP6 | 6.60 |
| MCM10 | 2.63 | ART3 | 3.24 | GPX3 | 5.82 | MUCL1 | 6.37 |
| GBP5 | 2.62 | ELF5 | 3.23 | CXCL14 | 5.51 | AR | 6.20 |
| EZH2 | 2.61 | MIA | 3.21 | ENPP2 | 5.32 | TOX3 | 5.90 |
| CCL5 | 2.60 | TTYH1 | 3.15 | SRPX | 5.27 | GPR160 | 5.32 |
| ADAMDEC1 | 2.55 | PTPRZ1 | 3.15 | DPT | 5.23 | PRR15 | 5.25 |
| CEP55 | 2.54 | COL9A3 | 3.08 | IL33 | 5.19 | FAM110C | 5.19 |
| Classification Methods | % Accuracy | ||||
|---|---|---|---|---|---|
| BLIA | BLIS | MES | LAR | ||
| SVM | Linear SVM | 92.3 ± 0.4 | 93.6 ± 0.5 | 94.8 ± 0.2 | 98.0 ± 0.3 |
| Quadratic SVM | 93.9 ± 0.7 | 93.4 ± 1.1 | 94.5 ± 0.2 | 98.4 ± 0.2 | |
| Cubic SVM | 94.2 ± 1.2 | 93.9 ± 1.0 | 94.4 ± 0.4 | 98.4 ± 0.2 | |
| Fine Gaussian SVM | 72.7 ± 0.0 | 69.7 ± 0.0 | 76.3 ± 0.0 | 81.3 ± 0.0 | |
| Medium Gaussian SVM | 95.7 ± 0.7 | 95.6 ± 0.7 | 95.0 ± 0.3 | 98.3 ± 0.2 | |
| Coarse Gaussian SVM | 74.2 ± 0.0 | 83.1 ± 0.4 | 91.4 ± 0.5 | 98.8 ± 0.2 | |
| KNN | Fine KNN | 89.0 ± 1.0 | 88.5 ± 0.7 | 93.3 ± 0.4 | 98.4 ± 0.2 |
| Medium KNN | 92.3 ± 0.8 | 93.4 ± 0.4 | 93.5 ± 0.4 | 97.8 ± 0.4 | |
| Coarse KNN | 72.7 ± 0.0 | 69.7 ± 0.0 | 76.3 ± 0.0 | 81.3 ± 0.0 | |
| Cosine KNN | 94.8 ± 0.8 | 94.3 ± 1.3 | 94.7 ± 0.6 | 98.5 ± 0.4 | |
| Cubic KNN | 92.1 ± 1.0 | 93.8 ± 0.7 | 93.9 ± 0.5 | 97.6 ± 0.8 | |
| Weighted KNN | 94.1 ± 0.8 | 93.7 ± 0.8 | 94.7 ± 0.5 | 98.1 ± 0.2 | |
| Ensemble | Boosted Trees | 72.7 ± 0.0 | 69.7 ± 0.0 | 76.3 ± 0.0 | 81.3 ± 0.0 |
| Bagged trees | 86.6 ± 1.8 | 88.5 ± 1.0 | 90.7 ± 1.4 | 92.9 ± 0.8 | |
| Subspace Discriminate | 91.1 ± 1.9 | 91.5 ± 1.1 | 92.2 ± 1.1 | 98.0 ± 0.5 | |
| Subspace KNN | 90.5 ± 1.0 | 90.3 ± 1.3 | 93.8 ± 0.2 | 98.4 ± 0.2 | |
| RUSBoosted Trees | 88.2 ± 1.8 | 88.9 ± 0.9 | 92.4 ± 0.7 | 95.4 ± 0.6 | |
| Tree | Fine Tree | 82.3 ± 2.1 | 79.0 ± 2.5 | 89.7 ± 1.6 | 95.5 ± 1.6 |
| Medium Tree | 81.7 ± 2.2 | 79.0 ± 2.5 | 89.7 ± 1.6 | 95.5 ± 1.6 | |
| Coarse Tree | 83.5 ± 1.7 | 80.9 ± 2.4 | 89.9 ± 1.8 | 95.5 ± 1.6 | |
| Linear Discriminant | Linear Discriminant | 86.0 ± 1.2 | 90.2 ± 0.8 | 90.2 ± 1.1 | 97.4 ± 0.6 |
| Logistic Regression | Logistic Regression | 64.7 ± 1.7 | 61.5 ± 2.9 | 61.9 ± 4.0 | 74.0 ± 3.4 |
| Naïve Bayes | Gaussian Naïve Bayes | 76.8 ± 0.8 | 78.4 ± 0.2 | 95.0 ± 0.5 | 97.9 ± 0.2 |
| Kernel Naïve Bayes | 83.0 ± 0.9 | 79.3 ± 0.4 | 95.0 ± 0.9 | 97.5 ± 0.6 | |
| BLIA | BLIS | MES | LAR | |
|---|---|---|---|---|
| Medium Gaussian SVM | Medium Gaussian SVM | Medium Gaussian SVM | Coarse Gaussian SVM | |
| Recall | 85.2 | 88.3 | 93.2 | 100.0 |
| Specificity | 99.3 | 97.1 | 96.1 | 98.8 |
| Precision (PPV) | 97.9 | 93.0 | 87.2 | 94.6 |
| Negative Predictive value | 94.7 | 95.0 | 98.0 | 100.0 |
| F1 score | 0.91 | 0.91 | 0.90 | 0.97 |
| Unclassified Samples | |
|---|---|
| Gene | FC |
| EN1 | 2.33 |
| PROM1 | 2.11 |
| CCL2 | 2.09 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bissanum, R.; Chaichulee, S.; Kamolphiwong, R.; Navakanitworakul, R.; Kanokwiroon, K. Molecular Classification Models for Triple Negative Breast Cancer Subtype Using Machine Learning. J. Pers. Med. 2021, 11, 881. https://doi.org/10.3390/jpm11090881
Bissanum R, Chaichulee S, Kamolphiwong R, Navakanitworakul R, Kanokwiroon K. Molecular Classification Models for Triple Negative Breast Cancer Subtype Using Machine Learning. Journal of Personalized Medicine. 2021; 11(9):881. https://doi.org/10.3390/jpm11090881
Chicago/Turabian StyleBissanum, Rassanee, Sitthichok Chaichulee, Rawikant Kamolphiwong, Raphatphorn Navakanitworakul, and Kanyanatt Kanokwiroon. 2021. "Molecular Classification Models for Triple Negative Breast Cancer Subtype Using Machine Learning" Journal of Personalized Medicine 11, no. 9: 881. https://doi.org/10.3390/jpm11090881