
Genetic Variants in PGE2 Receptors Modulate the Risk of Nephrosclerosis and Clinical Outcomes in These Patients

 ,
, 
Abstract
:1. Introduction
2. Results
2.1. Genetic Associations with the Risk of Nephrosclerosis
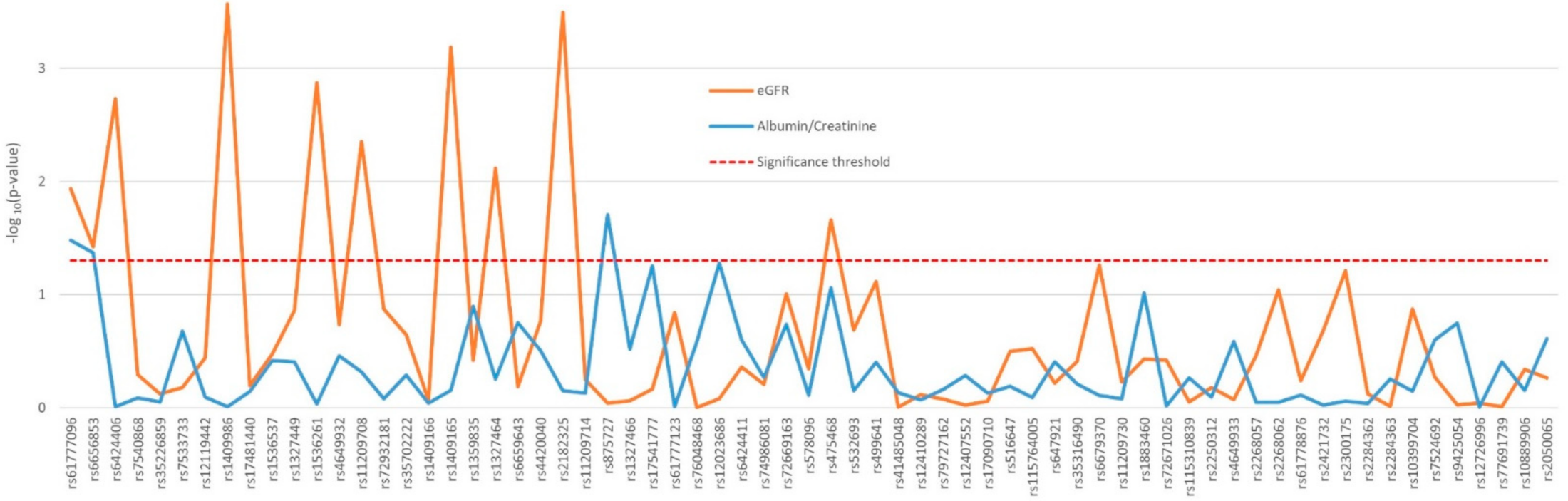
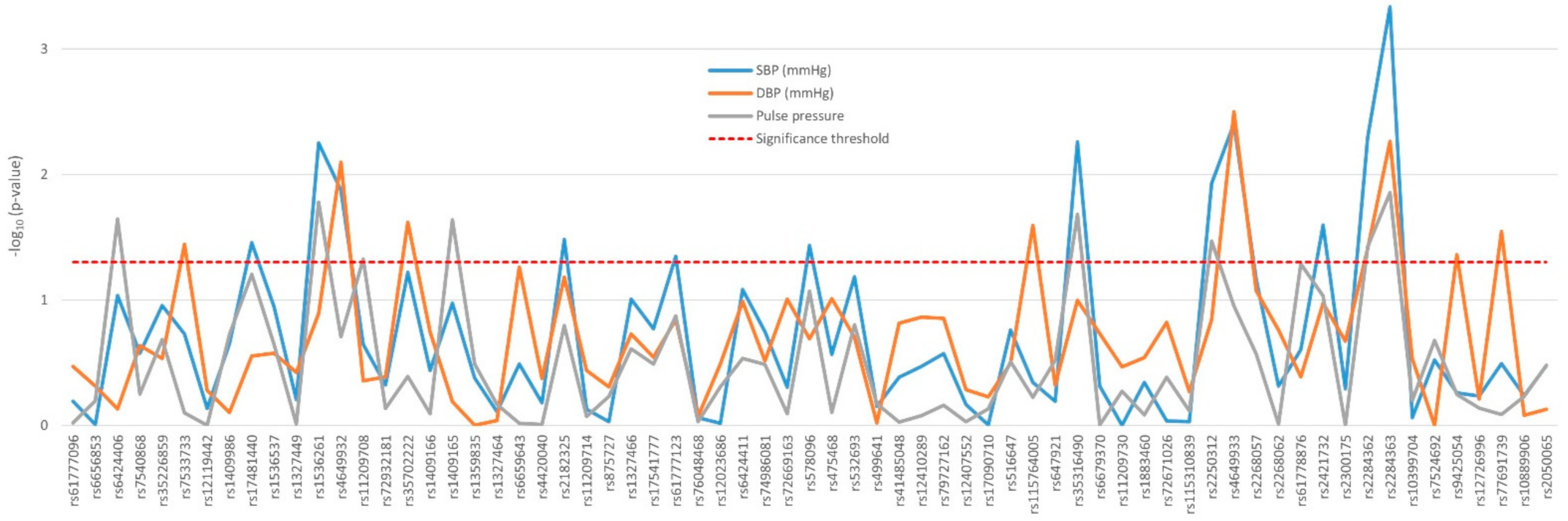
2.2. Impact of Polymorphisms on Renal Function and Blood Pressure Traits
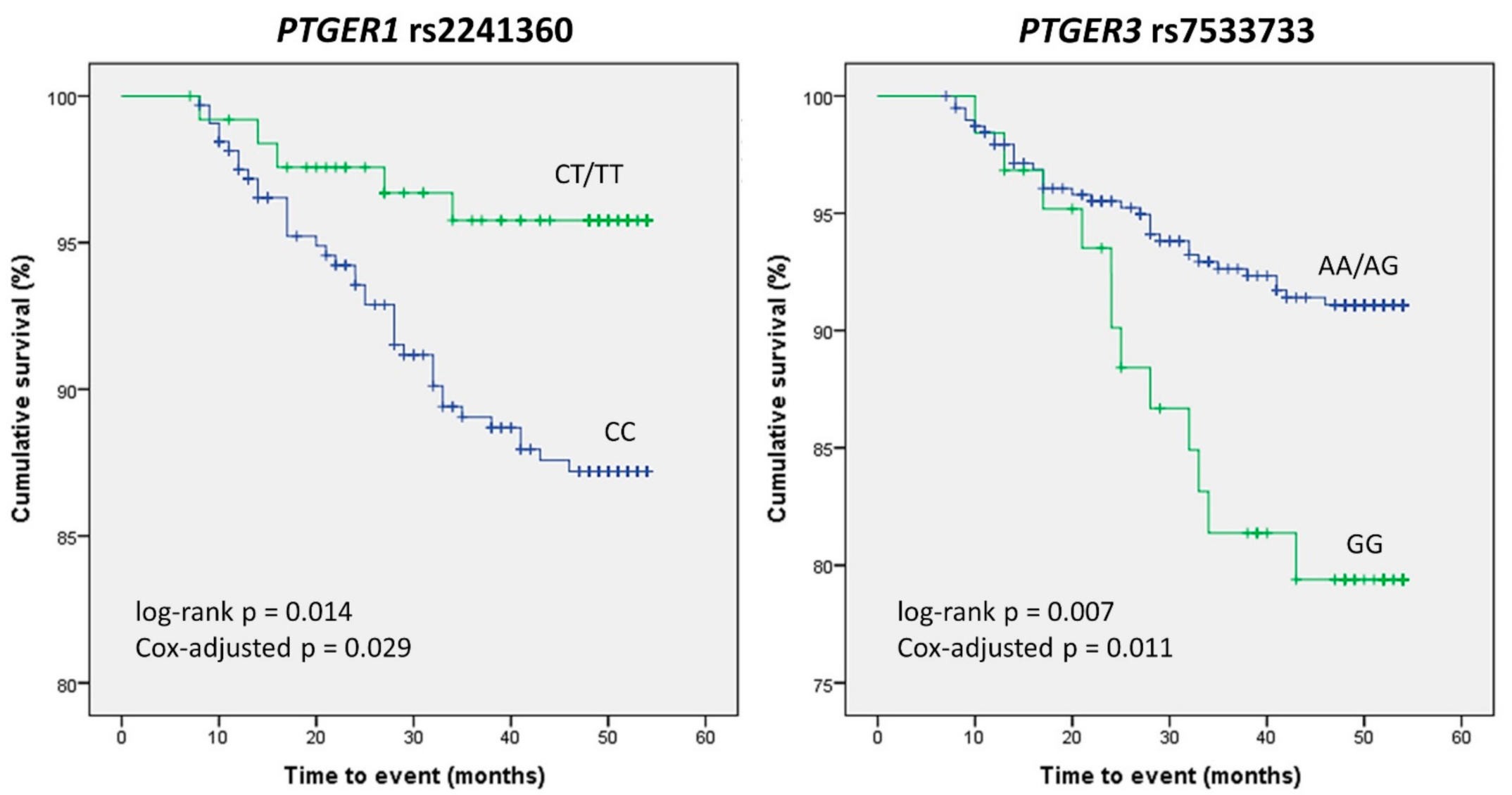
2.3. Association of PTGER Variants with the Incidence of Cardiovascular Events
3. Discussion
4. Patients and Methods
4.1. Study Design
4.2. SNP Selection and Genotype Analysis
4.3. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sever, M.Ş.; Jager, K.J.; Vanholder, R.; Stengel, B.; Harambat, J.; Finne, P.; Tesař, V.; Barbullushi, M.; Bumblytė, I.A.; Zakharova, E.; et al. A roadmap for optimizing chronic kidney disease patient care and patient-oriented research in the Eastern European nephrology community. Clin. Kidney J. 2021, 14, 23–35. [Google Scholar] [CrossRef]
- Foreman, K.J.; Marquez, N.; Dolgert, A.; Fukutaki, K.; Fullman, N.; McGaughey, M.; Pletcher, M.A.; Smith, A.E.; Tang, K.; Yuan, C.W.; et al. Forecasting life expectancy, years of life lost, and all-cause and cause-specific mortality for 250 causes of death: Reference and alternative scenarios for 2016–40 for 195 countries and territories. Lancet 2018, 392, 2052–2090. [Google Scholar] [CrossRef] [Green Version]
- Hill, G.S. Hypertensive nephrosclerosis. Curr. Opin. Nephrol. Hypertens. 2008, 17, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Foley, R.N.; Parfrey, P.S.; Sarnak, M.J. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am. J. Kidney Dis. 1998, 32, S112–S119. [Google Scholar] [CrossRef] [PubMed]
- Romero, J.C.; Reckelhoff, J.F. State-of-the-Art lecture. Role of angiotensin and oxidative stress in essential hypertension. Hypertension 1999, 34, 943–949. [Google Scholar] [CrossRef] [Green Version]
- Fanelli, A.; Ghisi, D.; Aprile, P.L.; Lapi, F. Cardiovascular and cerebrovascular risk with nonsteroidal anti-inflammatory drugs and cyclooxygenase 2 inhibitors: Latest evidence and clinical implications. Ther. Adv. Drug Saf. 2017, 8, 173–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diez, R.R.R.; Muñoz, A.T.; Orejudo, M.; Exposito, L.M.; Santos, L.; Mateos, S.R.; Navarro, E.C.; Santamaria, L.T.; Marchant, V.; Ortiz, A.; et al. Interleukin-17A: Possible mediator and therapeutic target in hypertension. Nefrologia 2021, 41, 244–257. [Google Scholar]
- O’Sullivan, E.D.; Hughes, J.; Ferenbach, D.A. Renal Aging: Causes and Consequences. J. Am. Soc. Nephrol. 2017, 28, 407–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francois, H.; Facemire, C.; Kumar, A.; Audoly, L.; Koller, B.; Coffman, T. Role of microsomal prostaglandin E synthase 1 in the kidney. J. Am. Soc. Nephrol. 2007, 18, 1466–1475. [Google Scholar] [CrossRef]
- Mohamed, R.; Jayakumar, C.; Ranganathan, P.V.; Ganapathy, V.; Ramesh, G. Kidney proximal tubular epithelial-specific overexpression of netrin-1 suppresses inflammation and albuminuria through suppression of COX-2-mediated PGE2 production in streptozotocin-induced diabetic mice. Am. J. Pathol. 2012, 181, 1991–2002. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.; Loutzenhiser, K.; Loutzenhiser, R. Biphasic actions of prostaglandin E(2) on the renal afferent arteriole: Role of EP(3) and EP(4) receptors. Circ. Res. 2000, 86, 663–670. [Google Scholar] [CrossRef] [Green Version]
- Nasrallah, R.; Hassouneh, R.; Hebert, R.L. Chronic kidney disease: Targeting prostaglandin E2 receptors. Am. J. Physiol. Renal. Physiol. 2014, 307, F243–F250. [Google Scholar] [CrossRef]
- Wuttke, M.; Kottgen, A. Insights into kidney diseases from genome-wide association studies. Nat. Rev. Nephrol. 2016, 12, 549–562. [Google Scholar] [CrossRef]
- Limou, S.; Vince, N.; Parsa, A. Lessons from CKD-Related Genetic Association Studies-Moving Forward. Clin. J. Am. Soc. Nephrol. 2018, 13, 140–152. [Google Scholar] [CrossRef] [Green Version]
- Sõber, S.; Org, E.; Kepp, K.; Juhanson, P.; Eyheramendy, S.; Gieger, C.; Lichtner, P.; Klopp, N.; Veldre, G.; Viigimaa, M.; et al. Targeting 160 candidate genes for blood pressure regulation with a genome-wide genotyping array. PLoS ONE 2009, 4, e6034. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Nakayama, T.; Soma, M.; Aoi, N.; Kosuge, K.; Haketa, A.; Izumi, Y.; Matsumoto, K.; Sato, N.; Kokubun, S. Association between prostaglandin E2 receptor gene and essential hypertension. Prostaglandins Leukot. Essent. Fat. Acids 2007, 77, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Szczeklik, W.; Sanak, M.; Rostoff, P.; Piwowarska, W.; Jakiela, B.; Szczeklik, A. Common polymorphisms of cyclooxygenase-2 and prostaglandin E2 receptor and increased risk for acute coronary syndrome in coronary artery disease. Thromb. Haemost. 2008, 100, 893–898. [Google Scholar] [CrossRef]
- Robles, N.R.; Fici, F.; Ari Bakir, E.; Grassi, G. Does established vascular kidney disease exist? J. Clin. Hypertens. (Greenwich) 2020, 22, 296–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robles, N.R.; Romero, B.; Bureo, N.; Macias, R.; Sanchez Casado, E.; Cubero, J.J. Hypertensive vascular disease: Evolution of its incidence in the period 1991-2007 and survival prior to end-stage renal disease. Nefrologia 2010, 30, 304–309. [Google Scholar] [PubMed]
- Hao, S.; DelliPizzi, A.; Quiroz-Munoz, M.; Jiang, H.; Ferreri, N.R. The EP3 receptor regulates water excretion in response to high salt intake. Am. J. Physiol. Renal. Physiol. 2016, 311, F822–F829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasrallah, R.; Hassouneh, R.; Hebert, R.L. PGE2, Kidney Disease, and Cardiovascular Risk: Beyond Hypertension and Diabetes. J. Am. Soc. Nephrol. 2016, 27, 666–676. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Wu, X.; Zeng, R.; Yin, Y.; Guo, T.; Xu, Y.; Zhang, Y.; Leng, J.; Ge, J.; Yu, G.; et al. Prostaglandin E2 sequentially activates E-prostanoid receptor-3 and thromboxane prostanoid receptor to evoke contraction and increase in resistance of the mouse renal vasculature. FASEB J. 2020, 34, 2568–2578. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Mitu, G.M.; Hirschberg, R. Osmotic polyuria: An overlooked mechanism in diabetic nephropathy. Nephrol. Dial. Transplant. 2008, 23, 2167–2172. [Google Scholar] [CrossRef] [PubMed]
- Audoly, L.P.; Ruan, X.; Wagner, V.A.; Goulet, J.L.; Tilley, S.L.; Koller, B.H.; Coffman, T.M.; Arendshorst, W.J. Role of EP(2) and EP(3) PGE(2) receptors in control of murine renal hemodynamics. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H327–H333. [Google Scholar] [CrossRef] [Green Version]
- Kvirkvelia, N.; McMenamin, M.; Chaudhary, K.; Bartoli, M.; Madaio, M.P. Prostaglandin E2 promotes cellular recovery from established nephrotoxic serum nephritis in mice, prosurvival, and regenerative effects on glomerular cells. Am. J. Physiol. Renal. Physiol. 2013, 304, F463–F470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweda, F.; Klar, J.; Narumiya, S.; Nusing, R.M.; Kurtz, A. Stimulation of renin release by prostaglandin E2 is mediated by EP2 and EP4 receptors in mouse kidneys. Am. J. Physiol. Renal. Physiol. 2004, 287, F427–F433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Guan, Y.; Schneider, A.; Brandon, S.; Breyer, R.M.; Breyer, M.D. Characterization of murine vasopressor and vasodepressor prostaglandin E(2) receptors. Hypertension 2000, 35, 1129–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swan, C.E.; Breyer, R.M. Prostaglandin E2 modulation of blood pressure homeostasis: Studies in rodent models. Prostaglandins Other Lipid Mediat. 2011, 96, 10–13. [Google Scholar] [CrossRef] [Green Version]
- Chopra, N.; Yang, T.; Asghari, P.; Moore, E.D.; Huke, S.; Akin, B.; Cattolica, R.A.; Perez, C.F.; Hlaing, T.; Ritschel, B.E.C.K.; et al. Ablation of triadin causes loss of cardiac Ca2+ release units, impaired excitation-contraction coupling, and cardiac arrhythmias. Proc. Natl. Acad. Sci. USA 2009, 106, 7636–7641. [Google Scholar] [CrossRef] [Green Version]
- Anzawa, R.; Seki, S.; Nagoshi, T.; Taniguchi, I.; Feuvray, D.; Yoshimura, M. The role of Na+/H+ exchanger in Ca2+ overload and ischemic myocardial damage in hearts from type 2 diabetic db/db mice. Cardiovasc. Diabetol. 2012, 11, 33. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Wang, G.; Liu, W.; Ding, W.; Dong, M.; Zheng, N.; Ye, H.; Liu, J. Hypoxia-Induced Mitogenic Factor Promotes Cardiac Hypertrophy via Calcium-Dependent and Hypoxia-Inducible Factor-1alpha Mechanisms. Hypertension 2018, 72, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.; Tilly, P.; Hentsch, D.; Vonesch, J.L.; Fabre, J.E. Vascular wall-produced prostaglandin E2 exacerbates arterial thrombosis and atherothrombosis through platelet EP3 receptors. J. Exp. Med. 2007, 204, 311–320. [Google Scholar] [CrossRef] [Green Version]
- Fabre, J.E.; Nguyen, M.; Athirakul, K.; Coggins, K.; McNeish, J.D.; Austin, S.; Parise, L.K.; FitzGerald, G.A.; Coffman, T.M.; Koller, B.H. Activation of the murine EP3 receptor for PGE2 inhibits cAMP production and promotes platelet aggregation. J. Clin. Investig. 2001, 107, 603–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.; Hara, A.; Xiao, C.Y.; Okada, Y.; Takahata, O.; Nakaya, K.; Sugimoto, Y.; Ichikawa, A.; Narumiya, S.; Ushikubi, F. Increased bleeding tendency and decreased susceptibility to thromboembolism in mice lacking the prostaglandin E receptor subtype EP(3). Circulation 2001, 104, 1176–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lannoy, M.; Valluru, M.K.; Chang, L.; Ali, F.A.; Peters, D.J.M.; Streets, A.J.; Ong, A.C.M. The positive effect of selective prostaglandin E2 receptor EP2 and EP4 blockade on cystogenesis in vitro is counteracted by increased kidney inflammation in vivo. Kidney Int. 2020, 98, 404–419. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, D.; Betriu, A.; Martinez-Alonso, M.; Vidal, T.; Valdivielso, J.M.; Fernandez, E. Observational multicenter study to evaluate the prevalence and prognosis of subclinical atheromatosis in a Spanish chronic kidney disease cohort: Baseline data from the NEFRONA study. BMC Nephrol. 2014, 15, 168. [Google Scholar] [CrossRef] [Green Version]
- Basilio, L.C.; Cortés, M.A.; Jerez, A.G.; Rodríguez, A.L.; Agudo, A.O.; Valdivielso, J.M.; Puyol, D.R.; Puyol, M.R. Quality Assurance of Samples and Processes in the Spanish Renal Research Network (REDinREN) Biobank. Biopreserv. Biobank 2016, 14, 499–510. [Google Scholar] [CrossRef]
- Valls, J.; Cambray, S.; Guallar, C.P.; Bozic, M.; López, M.B.; Fernández, E.; Betriu, À.; Rodríguez, I.; Valdivielso, J.M. Association of Candidate Gene Polymorphisms With Chronic Kidney Disease: Results of a Case-Control Analysis in the Nefrona Cohort. Front. Genet. 2019, 10, 118. [Google Scholar] [CrossRef] [Green Version]
- Mota-Zamorano, S.; Luna, E.; Garcia-Pino, G.; Gonzalez, L.M.; Gervasini, G. Combined donor-recipient genotypes of leptin receptor and adiponectin gene polymorphisms affect the incidence of complications after renal transplantation. Mol. Genet. Metab. Rep. 2020, 25, 100648. [Google Scholar] [CrossRef]
- Gervasini, G.; Pino, G.G.; Zamorano, S.M.; Luna, E.; Cerrada, M.G.; Tormo, M.Á.; Cubero, J.J. Association of polymorphisms in leptin and adiponectin genes with long-term outcomes in renal transplant recipients. Pharm. J. 2020, 20, 388–397. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Control (n = 716) | CKD 3 (n = 307) | CKD 4–5 (n = 123) | CKD 5D (n = 63) | Total (n = 1209) | p Value (Control vs. CKD) | p Value (between CKD Groups) | |
|---|---|---|---|---|---|---|---|
| Age (yrs) | 60 (51–68) | 67 (61–71) | 64 (57–71) | 61 (50–68) | 62 (54–69) | 6.2 × 10–17 | 1.05 × 10–4 |
| Males (%) | 359 (50.2) | 211 (68.7) | 80 (65.0) | 43 (68.3) | 693 (57.4) | 7.87 × 10–10 | 0.758 |
| Ethnicity | |||||||
| Caucasian | 709(99.0) | 304 (99.0) | 120 (97.6) | 58 (92.1) | 1191 (98.5) | 0.065 | 0.003 |
| Others | 7 (1.0) | 3 (1.0) | 3 (2.4) | 5 (7.9) | 18 (1.5) | ||
| Weight (kg) | 75.2 (66–85.8) | 80.3 (72–88.6) | 78.6 (67.5–87.2) | 75 (64.8–86) | 77.3 (67.6–86.7) | 1.1 × 10–5 | 0.030 |
| BMI | 30.4 (26.4–93) | 30.3 (27.5–34.6) | 28.8 (25.8–33.1) | 27 (24.2–31.1) | 30 (26.5–37.5) | 1.6 × 10–4 | 7.0 × 10–6 |
| Hypertension | 405 (56.6) | 293 (95.4) | 121 (98.4) | 60 (95.2) | 879 (72.8) | 1.95 × 10–61 | 0.333 |
| DM | 55 (7.7) | 74 (24.1) | 32 (26.0) | 8 (12.7) | 169 (14.0) | 4.23 × 10–14 | 0.100 |
| Hyperlipidemia | 175 (35.9) | 184 (67.2) | 91 (74.0) | 39 (61.9) | 489 (51.6) | 7.43 × 10–24 | 0.203 |
| Smoking | |||||||
| Non-smoker | 328 (45.8) | 124 (40.4) | 53 (43.1) | 26 (41.3) | 531 (43.9) | 0.255 | 0.929 |
| Current-smoker | 125 (17.5) | 53 (17.3) | 22 (17.9) | 13 (20.6) | 213 (17.6) | ||
| Former-smoker | 263 (36.7) | 130 (42.3) | 48 (39.0) | 24 (38.1) | 465 (38.5) | ||
| Systolic blood pressure (mmHg) | 136 (124–149) | 145 (132–160) | 146 (133.5–161) | 139 (130–155) | 139 (127–154) | 3.08 × 10–14 | 0.162 |
| Diastolic blood pressure (mmHg) | 81 (74–88) | 81 (75–89) | 79 (73–88) | 80 (70–90) | 81 (74–88.5) | 0.838 | 0.410 |
| Pulse pressure (mmHg) | 54 (46–64.5) | 64 (51–77) | 64 (55–76.5) | 60 (50–70) | 57 (48–70) | 5.6 × 10–20 | 0.123 |
| Total cholesterol (mg/dL) | 200.7 (180–222.8) | 185 (159.8–213.3) | 175.5 (148.5–203) | 160.5 (137.8–185.5) | 192 (165–216) | 1.9 × 10–17 | 1.9 × 10–5 |
| Cardiovascular events | 9 (1.3) | 25 (8.1) | 10 (8.1) | 6 (9.5) | 50 (4.1) | 1.33 × 10–9 | 0.933 |
| Creatinine (mg/dL) | 0.8 (0.7–1) | 1.5 (1.3–1.7) | 2.9 (2.5–3.6) | 1 (0.8–1.6) | 1.06 × 10–102 | 3.09 × 10–40 | |
| Albumin/creatinine (mg/g) | 6.4 (4–41.2) | 37.1 (7.5–195.1) | 212.1 (46–601) | 33.4 (5.1–182.4) | 1.27 × 10–11 | 3.6 × 10–5 | |
| eGFR (ml/min/1.73 m²) | 88.8 (78.7–100) | 45 (37.4–50.8) | 21 (16.2–25.6) | 71.2 (40.2–90.1) | 2.31 × 10–143 | 4.39 × 10–56 | |
| Polymorphism | Genotype | Control n (%) | CKD n (%) | OR | p Value |
|---|---|---|---|---|---|
| rs11209708 | A/A | 558 (80.5) | 369 (77.4) | 1.41 (1.01–1.97) | 0.043 |
| A/G-G/G | 135 (19.5) | 108 (22.6) | |||
| rs6424411 | A/A | 330 (48.0) | 203 (42.7) | 1.32 (1.01–1.74) | 0.045 |
| A/G-G/G | 358 (52.0) | 272 (57.3) | |||
| rs475468 | G/G | 254 (37.0) | 207 (43.1) | 0.72 (0.54–0.95) | 0.018 |
| G/A-A/A | 432 (63.0) | 273 (56.9) | |||
| rs6679370 | A/A | 201 (29.1) | 166 (34.4) | 0.71 (0.53–0.95) | 0.022 |
| A/G-G/G | 490 (70.9) | 316 (65.6) | |||
| rs11209730 | A/A | 475 (66.6) | 306 (62.7) | 1.38 (1.04–1.83) | 0.024 |
| A/G-G/G | 238 (33.4) | 182 (37.3) | |||
| rs2250312 | C/C | 311 (43.7) | 191 (38.9) | 1.41 (1.07–1.84) | 0.013 |
| C/T-T/T | 401 (56.3) | 300 (61.1) | |||
| rs2268062 | T/T | 209 (29.3) | 171 (34.9) | 0.69 (0.51–0.92) | 0.011 |
| T/C-C/C | 504 (70.7) | 319 (65.1) | |||
| rs61778876 | A/A | 646 (93.4) | 460 (95.4) | 0.54 (0.31–0.95) | 0.03 |
| A/G | 46 (6.6) | 22 (4.6) | |||
| rs2300175 | C/C | 207 (29.0) | 170 (34.7) | 0.69 (0.52–0.92) | 0.012 |
| C/T-T/T | 507 (71.0) | 320 (65.3) | |||
| rs10399704 | G/G | 163 (23.4) | 139 (29.2) | 0.72 (0.52–0.98) | 0.035 |
| G/A-A/A | 534 (76.6) | 337 (70.8) |
| B | SE | Wald | OR | CI | p Value | |
|---|---|---|---|---|---|---|
| Sex | 0.748 | 0.15 | 25.87 | 2.11 | (1.58–2.82) | 3.65 × 10–7 |
| Diabetes | 1.119 | 0.21 | 27.39 | 3.06 | (2.01–4.65) | 1.66 × 10–7 |
| Hypertension | 3.077 | 0.27 | 127.95 | 21.70 | (12.73–36.98) | 1.15 × 10–29 |
| rs11209708 A/G-G/G | 0.348 | 0.18 | 3.75 | 1.42 | (1.00–2.01) | 0.053 |
| rs6424411 A/G-G/G | 0.251 | 0.15 | 2.95 | 1.29 | (0.97–1.71) | 0.086 |
| rs11209730 A/G-G/G | 0.37 | 0.15 | 5.83 | 1.45 | (1.07–1.95) | 0.016 |
| rs10399704 G/A-A/A | −0.345 | 0.17 | 4.16 | 0.71 | (0.51–0.99) | 0.041 |
| Constant | −3.573 | 0.34 | 111.52 | 0.03 | 4.54 × 10–26 |
| Variable | No CVE | CVE | p Value |
|---|---|---|---|
| Age (years) | 66 (59–71) | 67 (64–72.5) | 0.014 |
| Sex (male) | 300 (66.4) | 34 (82.9) | 0.019 |
| Ethnicity | |||
| Caucasian | 441 (97.6) | 41 (100) | 0.381 |
| Others | 11 (2.4) | 0 | |
| Weight | 79.8 (69.7–88) | 81.5 (70.9–91.5) | 0.568 |
| BMI (kg/m²) | 29.8 (26.5–33.4) | 29.7 (26.1–33) | 0.923 |
| Hypertension | 435 (96.2) | 39 (95.1) | 0.480 |
| DM | 100 (22.1) | 14 (34.1) | 0.064 |
| Hyperlipidemia | * 284 (67.8) | 30 (73.2) | 0.302 |
| Smoking | |||
| Non-smoker | 190 (42.0) | 13 (31.7) | 0.224 |
| Current-smoker | 77 (17.0) | 11 (26.8) | |
| Former-smoker | 185 (49.9) | 17 (41.5) | |
| Pulse pressure (mmHg) | 62 (51–75.4) | 67 (57.3–78.3) | 0.096 |
| Systolic lood pressure (mmHg) | 144 (132–160) | 145 (134–162) | 0.642 |
| Dyastolic blood pressure (mmHg) | 81 (74–89) | 77.5 (70.8–88.8) | 0.245 |
| Chronic kidney disease stage | |||
| Stage 3 | 282 (62.4) | 25 (61.0) | 0.933 |
| Stage 4–5 | 113 (25.0) | 10 (24.4) | |
| Dialysis | 57 12.6) | 6 (14.6) | |
| Total cholesterol (mg/dL) | 181 (154.7–209) | 165 (139.5–212) | 0.065 |
| Creatinine (mg/dL) | 1.7 (1.4–2.4) | 1.7 (1.3–2.3) | 0.689 |
| Albumin/Cr (mg/g) | 68.9 (9.9–294.3) | 173.6 (25.5–470.6) | 0.171 |
| eGFR (mL/min/1.73 m²) | 38.2 (26.6–47.7) | 37.4 (25–47) | 0.705 |
| Glucose (mg/dL) | 100.5 (90–114) | 102 (96.5–123.5) | 0.134 |
| Calcium (mg/dL) | 9.4 (9.1–9.7) | 9.5 (9.1–9.7) | 0.947 |
| Sodium (mEq/L) | 141 (139–142) | 141 (139–142) | 0.951 |
| Potassium (mEq/L) | 4.7 (4.4–5.1) | 4.8 (4.4–5) | 0.946 |
| Gene | Polymorphism | Model | Genotype | No CVE | CVE | OR | p Value |
|---|---|---|---|---|---|---|---|
| PTGER1 | rs2241360 | Dominant | CC | 316 (70.9) | 34 (85.0) | 0.43 (0.18–1.05) | 0.037 |
| CT/TT | 130 (29.1) | 6 (15.0) | |||||
| PTGER3 | rs7533733 | Recessive | AG/AA | 390 (86.5) | 29 (70.7) | 2.65 (1.28–5.46) | 0.010 |
| GG | 61 (13.5) | 12 (29.3) | |||||
| PTGER3 | rs12119442 | Dominant | G/G | 304 (69.7) | 21 (51.2) | 2.19 (1.15–4.18) | 0.014 |
| G/A-A/A | 132 (30.3) | 20 (48.8) | |||||
| PTGER3 | rs74986081 | Dominant | GG | 333 (75.7) | 34 (89.5) | 0.37 (0.13–1.05) | 0.035 |
| GA/AA | 107 (24.3) | 4 (10.5) | |||||
| PTGER3 | rs2268057 | Overdominant | CC/TT | 254 (56.3) | 16 (39.0) | 2.02 (1.05–3.88) | 0.025 |
| CT | 197 (43.7) | 25 (61.0) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González, L.M.; Robles, N.R.; Mota-Zamorano, S.; Valdivielso, J.M.; López-Gómez, J.; Gervasini, G. Genetic Variants in PGE2 Receptors Modulate the Risk of Nephrosclerosis and Clinical Outcomes in These Patients. J. Pers. Med. 2021, 11, 772. https://doi.org/10.3390/jpm11080772
González LM, Robles NR, Mota-Zamorano S, Valdivielso JM, López-Gómez J, Gervasini G. Genetic Variants in PGE2 Receptors Modulate the Risk of Nephrosclerosis and Clinical Outcomes in These Patients. Journal of Personalized Medicine. 2021; 11(8):772. https://doi.org/10.3390/jpm11080772
Chicago/Turabian StyleGonzález, Luz María, Nicolás Roberto Robles, Sonia Mota-Zamorano, José Manuel Valdivielso, Juan López-Gómez, and Guillermo Gervasini. 2021. "Genetic Variants in PGE2 Receptors Modulate the Risk of Nephrosclerosis and Clinical Outcomes in These Patients" Journal of Personalized Medicine 11, no. 8: 772. https://doi.org/10.3390/jpm11080772