
Efficient Genetic Safety Switches for Future Application of iPSC-Derived Cell Transplants

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids
2.1.1. AAVS1 Donor Plasmids
2.1.2. Lentiviral Vectors
2.2. Cell Cultivation
2.3. TALEN induced AAVS1 Safe Harbor Targeting
2.4. Verification of Correctly Targeted iPSC
2.5. Production of Lentiviral Particles
2.6. Transduction of iPSCs with Lentiviral Vectors
2.7. Embryoid Body-Based Differentiation into Macrophages
2.8. Cytospin Analysis
2.9. Flow Cytometry
2.10. RT qPCR
2.11. Vector Copy Number (VCN) Determination
2.12. In Vitro Ablation of Transgenic Cells
2.13. Animals
2.14. In Vivo Ablation of Transgenic Cells
2.15. Statistical Analyses
3. Results
3.1. Establishment of iPSC Lines Stably Expressig Safety Switches
3.2. Efficient Ablation of TK.007-Transgenic iPSC upon GCV Treatment
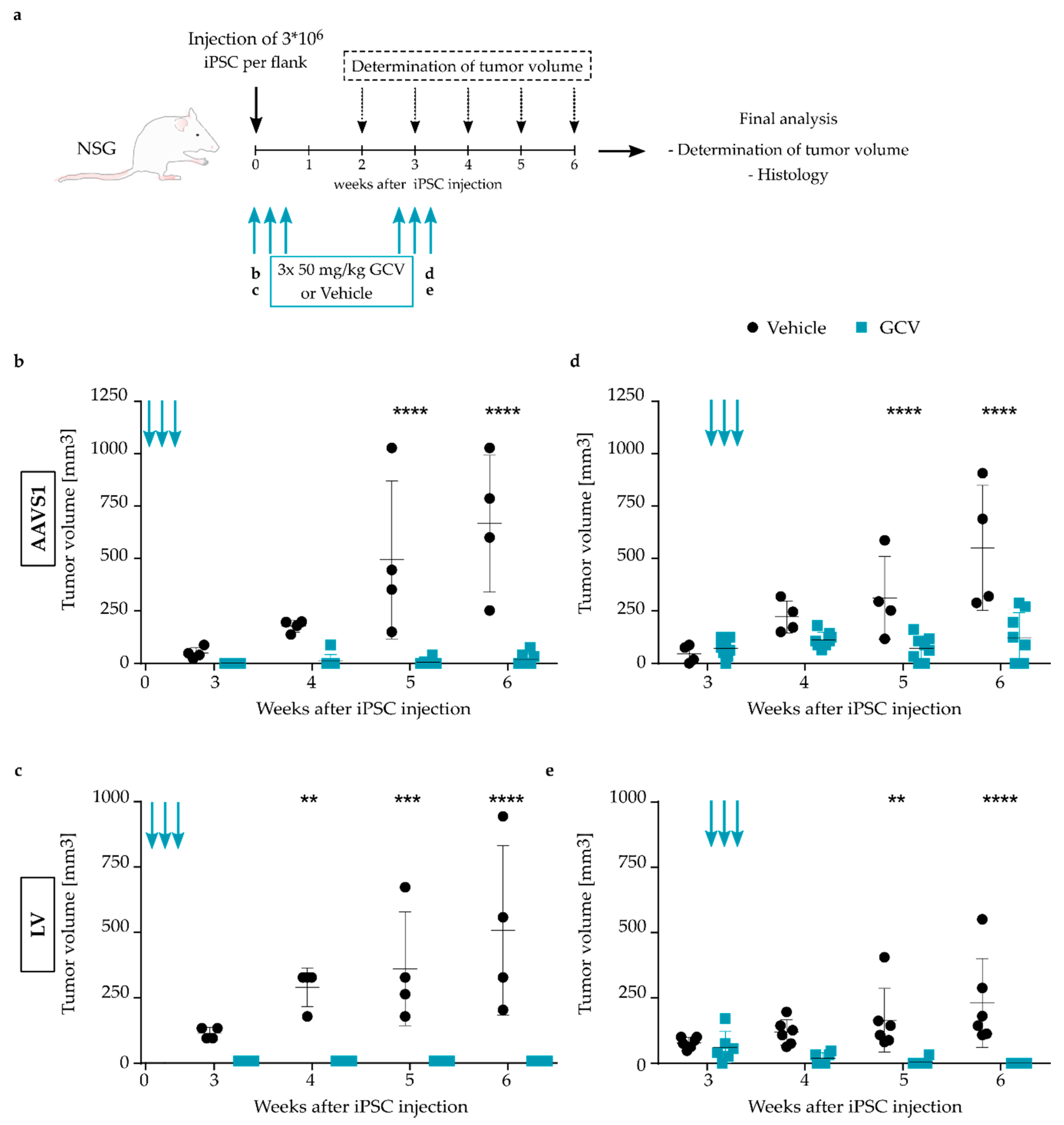
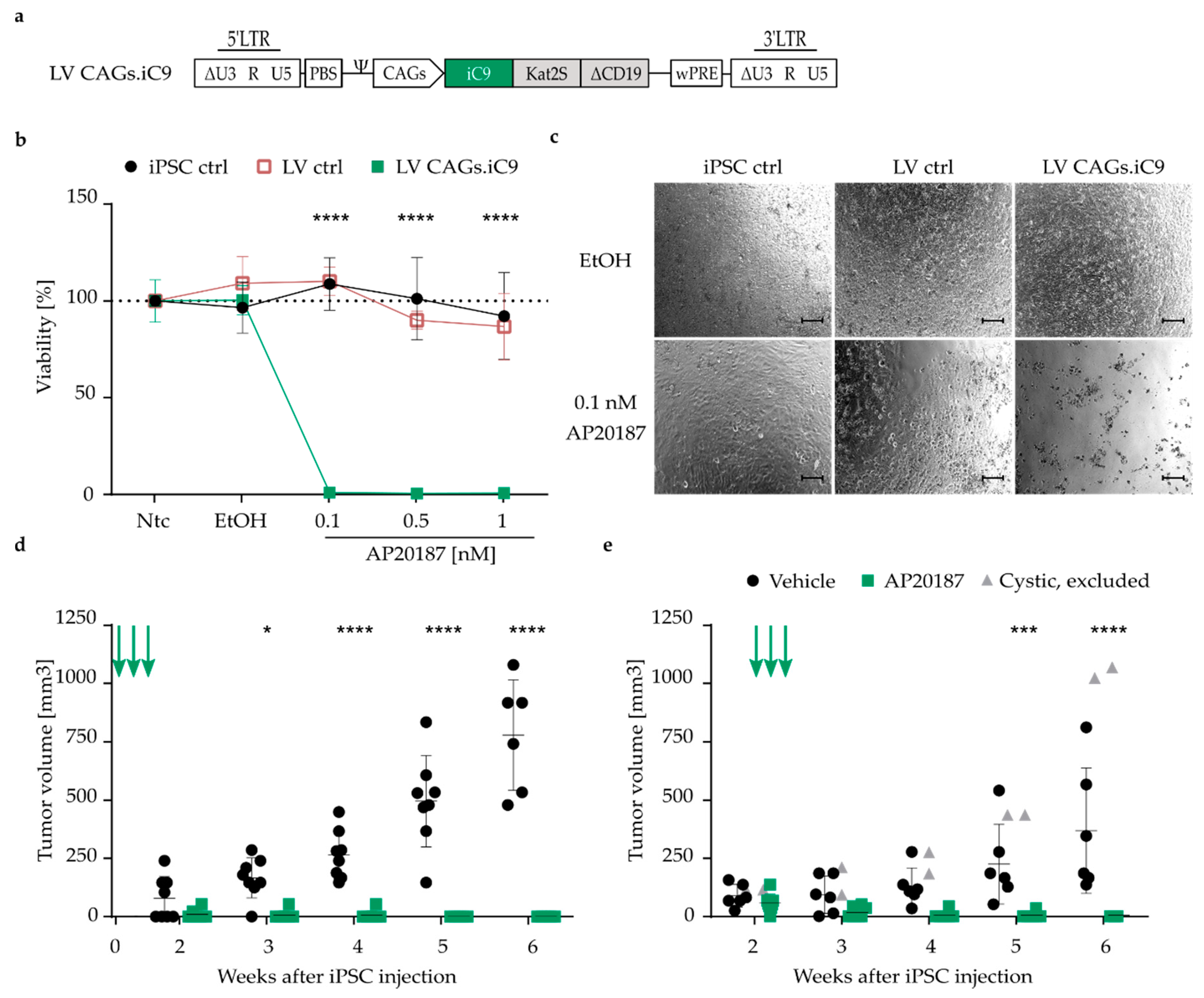
3.3. Highly Efficient iC9 Safety Switch Eliminates Teratomas In Vivo
3.4. Pluripotency-Specific Promoters Restrict the Effect of the Suicide Switch to iPSCs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandai, M.; Watanabe, A.; Kurimoto, Y.; Hirami, Y.; Morinaga, C.; Daimon, T.; Fujihara, M.; Akimaru, H.; Sakai, N.; Shibata, Y.; et al. Autologous Induced Stem-Cell–Derived Retinal Cells for Macular Degeneration. N. Engl. J. Med. 2017, 376, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Bloor, A.J.C.; Patel, A.; Griffin, J.E.; Gilleece, M.H.; Radia, R.; Yeung, D.T.; Drier, D.; Larson, L.S.; Uenishi, G.I.; Hei, D.; et al. Production, safety and efficacy of iPSC-derived mesenchymal stromal cells in acute steroid-resistant graft versus host disease: A phase I, multicenter, open-label, dose-escalation study. Nat. Med. 2020, 26, 1720–1725. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, J. iPS cell-based therapy for Parkinson’s disease: A Kyoto trial. Regen. Ther. 2020, 13, 18–22. [Google Scholar] [CrossRef]
- Lee, A.S.; Tang, C.; Rao, M.S.; Weissman, I.L.; Wu, J.C. Tumorigenicity as a Clinical Hurdle for Pluripotent Stem Cell Therapies. Nat. Med. 2013, 19, 998–1004. [Google Scholar] [CrossRef] [Green Version]
- Marin, V.; Cribioli, E.; Philip, B.; Tettamanti, S.; Pizzitola, I.; Biondi, A.; Biagi, E.; Pule, M. Comparison of different suicide-gene strategies for the safety improvement of genetically manipulated T cells. Hum. Gene Ther. Methods 2012, 23, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Di Stasi, A.; Tey, S.-K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [Green Version]
- Chalmers, D.; Ferrand, C.; Apperley, J.F.; Melo, J.V.; Ebeling, S.; Newton, I.; Duperrier, A.; Hagenbeek, A.; Garrett, E.; Tiberghien, P.; et al. Elimination of the truncated message from the herpes simplex virus thymidine kinase suicide gene. Mol. Ther. 2001, 4, 146–148. [Google Scholar] [CrossRef]
- Balzarini, J.; Liekens, S.; Solaroli, N.; El Omari, K.; Stammers, D.K.; Karlsson, A. Engineering of a single conserved amino acid residue of herpes simplex virus type 1 thymidine kinase allows a predominant shift from pyrimidine to purine nucleoside phosphorylation. J. Biol. Chem. 2006, 281, 19273–19279. [Google Scholar] [CrossRef] [Green Version]
- Preuß, E.; Treschow, A.; Newrzela, S.; Brücher, D.; Weber, K.; Felldin, U.; Alici, E.; Gahrton, G.; Von Laer, D.; Dilber, M.S.; et al. A Novel, Codon-Optimized HSVtk(A168H) Mutant for Suicide Gene Therapy. Hum. Gene Ther. 2010, 21, 929–941. [Google Scholar] [CrossRef]
- Preuß, E.; Muik, A.; Weber, K.; Otte, J.; Von Laer, D.; Fehse, B. Cancer suicide gene therapy with TK.007: Superior killing efficiency and bystander effect. J. Mol. Med. 2011, 89, 1113–1124. [Google Scholar] [CrossRef]
- Zhou, X.; Dotti, G.; Krance, R.A.; Martinez, C.A.; Naik, S.; Kamble, R.T.; Durett, A.G.; Dakhova, O.; Savoldo, B.; Di Stasi, A.; et al. Inducible caspase-9 suicide gene controls adverse effects from alloreplete T cells after haploidentical stem cell transplantation. Blood 2015, 125, 4103–4113. [Google Scholar] [CrossRef] [Green Version]
- Papapetrou, E.P.; Schambach, A. Gene insertion into genomic safe harbors for human gene therapy. Mol. Ther. 2016, 24, 678–684. [Google Scholar] [CrossRef] [Green Version]
- Kotin, R.M.; Siniscalco, M.; Samulski, R.J.; Zhu, X.D.; Hunter, L.; Laughlin, C.A.; McLaughlin, S.; Muzyczka, N.; Rocchi, M.; Berns, K.I. Site-specific integration by adeno-associated virus. Proc. Natl. Acad. Sci. USA 1990, 87, 2211–2215. [Google Scholar] [CrossRef] [Green Version]
- Oceguera-Yanez, F.; Kim, S.-I.; Matsumoto, T.; Tan, G.W.; Xiang, L.; Hatani, T.; Kondo, T.; Ikeya, M.; Yoshida, Y.; Inoue, H.; et al. Engineering the AAVS1 locus for consistent and scalable transgene expression in human iPSCs and their differentiated derivatives. Methods 2016, 101, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Wang, H.; Kiani, S.; Lai, C.S.; Gao, Q.; Cassady, J.P.; Cost, G.J.; Zhang, L.; Santiago, Y.; Miller, J.C.; et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nat. Biotechnol. 2011, 29, 731–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sens, M.J.; Hoffmann, D.; Lange, L.; Barbosa, P.V.; Morgan, M.A.; Falk, C.S.; Schambach, A. Knockout-Induced Pluripotent Stem Cells for Disease and Therapy Modeling of IL-10-Associated Primary Immunodeficiencies. Hum. Gene Ther. 2021, 32, 77–95. [Google Scholar] [CrossRef] [PubMed]
- Klatt, D.; Cheng, E.; Hoffmann, D.; Santilli, G.; Thrasher, A.J.; Brendel, C.; Schambach, A. Differential Transgene Silencing of Myeloid-Specific Promoters in the AAVS1 Safe Harbor Locus of Induced Pluripotent Stem Cell-Derived Myeloid Cells. Hum. Gene Ther. 2020, 31, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Ordovás, L.; Boon, R.; Pistoni, M.; Chen, Y.; Wolfs, E.; Guo, W.; Sambathkumar, R.; Bobis-Wozowicz, S.; Helsen, N.; Vanhove, J.; et al. Efficient recombinase-mediated cassette exchange in hPSCs to study the hepatocyte lineage reveals AAVS1 locus-mediated transgene inhibition. Stem Cell Rep. 2015, 5, 918–931. [Google Scholar] [CrossRef] [Green Version]
- Hacein-Bey-Abina, S.; Von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. LMO2-Associated Clonal T Cell Proliferation in Two Patients after Gene Therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef]
- Kaufmann, K.B.; Büning, H.; Galy, A.; Schambach, A.; Grez, M. Gene therapy on the move. EMBO Mol. Med. 2013, 5, 1642–1661. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Hong, S.G.; Winkler, T.; Spencer, D.M.; Jares, A.; Ichwan, B.; Nicolae, A.; Guo, V.; Larochelle, A.; Dunbar, C.E. Development of an inducible caspase-9 safety switch for pluripotent stem cell–based therapies. Mol. Ther. Methods Clin. Dev. 2014, 1, 14053. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, D.; Schott, J.W.; Geis, F.K.; Lange, L.; Müller, F.-J.; Lenz, D.; Zychlinski, D.; Steinemann, D.; Morgan, M.; Moritz, T.; et al. Detailed comparison of retroviral vectors and promoter configurations for stable and high transgene expression in human induced pluripotent stem cells. Gene Ther. 2017, 24, 298–307. [Google Scholar] [CrossRef]
- Hockemeyer, D.; Soldner, F.; Beard, C.; Gao, Q.; Mitalipova, M.; DeKelver, R.C.; Katibah, G.E.; Amora, R.; Boydston, E.A.; Zeitler, B.; et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 2009, 27, 851–857. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Liu, C.; Cerbini, T.; San, H.; Lin, Y.; Chen, G.; Rao, M.S.; Zou, J. Stable Enhanced Green Fluorescent Protein Expression After Differentiation and Transplantation of Reporter Human Induced Pluripotent Stem Cells Generated by AAVS1 Transcription Activator-Like Effector Nucleases. Stem Cells Transl. Med. 2014, 3, 821–835. [Google Scholar] [CrossRef]
- Hotta, A.; Cheung, A.Y.L.; Farra, N.; Vijayaragavan, K.; Seguin, C.; Draper, J.S.; Pasceri, P.; Maksakova, I.A.; Mager, D.L.; Rossant, J.; et al. Isolation of human iPS cells using EOS lentiviral vectors to select for pluripotency. Nat. Methods 2009, 6, 370–376. [Google Scholar] [CrossRef]
- Hotta, A.; Cheung, A.Y.L.; Farra, N.; Garcha, K.; Chang, W.Y.; Pasceri, P.; Stanford, W.L.; Ellis, J. EOS lentiviral vector selection system for human induced pluripotent stem cells. Nat. Protoc. 2009, 4, 1828–1844. [Google Scholar] [CrossRef]
- Vega-Crespo, A.; Truong, B.; Hermann, K.J.; Awe, J.P.; Chang, K.M.; Lee, P.C.; Schoenberg, B.E.; Wu, L.; Byrne, J.A.; Lipshutz, G.S. Investigating the functionality of an OCT4-short response element in human induced pluripotent stem cells. Mol. Ther. Methods Clin. Dev. 2016, 3, 16050. [Google Scholar] [CrossRef] [Green Version]
- Bedel, A.; Béliveau, F.; Lamrissi-Garcia, I.; Rousseau, B.; Moranvillier, I.; Rucheton, B.; Guyonnet-Dupérat, V.; Cardinaud, B.; De Verneuil, H.; Moreau-Gaudry, F.; et al. Preventing Pluripotent Cell Teratoma in Regenerative Medicine Applied to Hematology Disorders. Stem Cells Transl. Med. 2017, 6, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Sułkowski, M.; Konieczny, P.; Chlebanowska, P.; Majka, M. Introduction of exogenous HSV-TK suicide gene increases safety of keratinocyte-derived induced pluripotent stem cells by providing genetic “emergency exit” switch. Int. J. Mol. Sci. 2018, 19, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipus, A.; Janosz, E.; Ackermann, M.; Hetzel, M.; Dahlke, J.; Buchegger, T.; Wunderlich, S.; Martin, U.; Cathomen, T.; Schambach, A.; et al. Targeted integration of inducible caspase-9 in human iPSCs allows efficient in vitro clearance of iPSCs and iPSC-macrophages. Int. J. Mol. Sci. 2020, 21, 2481. [Google Scholar] [CrossRef] [Green Version]
- Merkert, S.; Wunderlich, S.; Bednarski, C.; Beier, J.; Haase, A.; Dreyer, A.-K.; Schwanke, K.; Meyer, J.; Göhring, G.; Cathomen, T.; et al. Efficient designer nuclease-based homologous recombination enables direct PCR screening for footprintless targeted human pluripotent stem cells. Stem Cell Rep. 2014, 2, 107–118. [Google Scholar] [CrossRef] [Green Version]
- Luker, K.; Pata, P.; Shemiakina, I.; Pereverzeva, A.; Stacer, A.; Shcherbo, D.; Pletnev, V.; Skolnaja, M.; Lukyanov, K.; Luker, G.; et al. Comparative study reveals better far-red fluorescent protein for whole body imaging. Sci. Rep. 2015, 5, 10332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luke, G.A.; de Felipe, P.; Lukashev, A.; Kallioinen, S.E.; Bruno, E.A.; Ryan, M.D. Occurrence, function and evolutionary origins of “2A-like” sequences in virus genomes. J. Gen. Virol. 2008, 89, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J.; Nguyen, M.; Trono, D.; Naldini, L. A Third-Generation Lentivirus Vector with a Conditional Packaging System. J. Virol. 1998, 72, 8463–8471. [Google Scholar] [CrossRef] [Green Version]
- Müller-Kuller, U.; Ackermann, M.; Kolodziej, S.; Brendel, C.; Fritsch, J.; Lachmann, N.; Kunkel, H.; Lausen, J.; Schambach, A.; Moritz, T.; et al. A minimal ubiquitous chromatin opening element (UCOE) effectively prevents silencing of juxtaposed heterologous promoters by epigenetic remodeling in multipotent and pluripotent stem cells. Nucleic Acids Res. 2015, 43, 1577–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schambach, A.; Bohne, J.; Baum, C.; Hermann, F.G.; Egerer, L.; von Laer, D.; Giroglou, T. Woodchuck hepatitis virus post-transcriptional regulatory element deleted from X protein and promoter sequences enhances retroviral vector titer and expression. Gene Ther. 2006, 13, 641–645. [Google Scholar] [CrossRef] [Green Version]
- Lachmann, N.; Happle, C.; Ackermann, M.; Lüttge, D.; Wetzke, M.; Merkert, S.; Hetzel, M.; Kensah, G.; Jara-Avaca, M.; Mucci, A.; et al. Gene correction of human induced pluripotent stem cells repairs the cellular phenotype in pulmonary alveolar proteinosis. Am. J. Respir. Crit. Care Med. 2014, 189, 167–182. [Google Scholar] [CrossRef]
- Mussolino, C.; Alzubi, J.; Fine, E.J.; Morbitzer, R.; Cradick, T.J.; Lahaye, T.; Bao, G.; Cathomen, T. TALENs facilitate targeted genome editing in human cells with high specificity and low cytotoxicity. Nucleic Acids Res. 2014, 42, 6762–6773. [Google Scholar] [CrossRef] [Green Version]
- Galla, M.; Schambach, A.; Towers, G.J.; Baum, C. Cellular Restriction of Retrovirus Particle-Mediated mRNA Transfer. J. Virol. 2008, 82, 3069–3077. [Google Scholar] [CrossRef] [Green Version]
- Lachmann, N.; Ackermann, M.; Frenzel, E.; Liebhaber, S.; Brennig, S.; Happle, C.; Hoffmann, D.; Klimenkova, O.; Lüttge, D.; Buchegger, T.; et al. Large-scale hematopoietic differentiation of human induced pluripotent stem cells provides granulocytes or macrophages for cell replacement therapies. Stem Cell Rep. 2015, 4, 282–296. [Google Scholar] [CrossRef] [Green Version]
- Schott, J.W.; Hoffmann, D.; Maetzig, T.; Müller, F.J.; Steinemann, D.; Zychlinski, D.; Cantz, T.; Baum, C.; Schambach, A. Improved retroviral episome transfer of transcription factors enables sustained cell fate modification. Gene Ther. 2014, 21, 938–949. [Google Scholar] [CrossRef] [PubMed]
- Philipp, F.; Selich, A.; Rothe, M.; Hoffmann, D.; Rittinghausen, S.; Morgan, M.A.; Klatt, D.; Glage, S.; Lienenklaus, S.; Neuhaus, V.; et al. Human Teratoma-Derived Hematopoiesis Is a Highly Polyclonal Process Supported by Human Umbilical Vein Endothelial Cells. Stem Cell Rep. 2018, 11, 1051–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naujok, O.; Kaldrack, J.; Taivankhuu, T.; Jörns, A.; Lenzen, S. Selective Removal of Undifferentiated Embryonic Stem Cells from Differentiation Cultures Through HSV1 Thymidine Kinase and Ganciclovir Treatment. Stem Cell Rev. Rep. 2010, 6, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Ciceri, F.; Bonini, C.; Lupo-Stanghellini, M.T.; Bondanza, A.; Traversari, C.; Salomoni, M.; Turchetto, L.; Colombi, S.; Bernardi, M.; Peccatori, J.; et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): A non-randomised phase I–II study. Lancet Oncol. 2009, 10, 489–500. [Google Scholar] [CrossRef]
- Bonini, C.; Bondanza, A.; Perna, S.K.; Kaneko, S.; Traversari, C.; Ciceri, F.; Bordignon, C. The suicide gene therapy challenge: How to improve a successful gene therapy approach. Mol. Ther. 2007, 15, 1248–1252. [Google Scholar] [CrossRef]
- Li, P.; Zhou, L.; Zhao, T.; Liu, X.; Zhang, P.; Liu, Y.; Zheng, X.; Li, Q. Caspase-9: Structure, mechanisms and clinical application. Oncotarget 2017, 8, 23996–24008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klatt, D.; Cheng, E.; Philipp, F.; Selich, A.; Dahlke, J.; Schmidt, R.E.; Schott, J.W.; Büning, H.; Hoffmann, D.; Thrasher, A.J.; et al. Targeted Repair of p47-CGD in iPSCs by CRISPR/Cas9: Functional Correction without Cleavage in the Highly Homologous Pseudogenes. Stem Cell Rep. 2019, 13, 590–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreyer, A.-K.; Hoffmann, D.; Lachmann, N.; Ackermann, M.; Steinemann, D.; Timm, B.; Siler, U.; Reichenbach, J.; Grez, M.; Moritz, T.; et al. TALEN-mediated functional correction of X-linked chronic granulomatous disease in patient-derived induced pluripotent stem cells. Biomaterials 2015, 69, 191–200. [Google Scholar] [CrossRef]
- Gornalusse, G.G.; Hirata, R.K.; Funk, S.E.; Riolobos, L.; Lopes, V.S.; Manske, G.; Prunkard, D.; Colunga, A.; Hanafi, L.-A.; Clegg, V.S.L.D.O.; et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells HHS Public Access. Nat. Biotechnol. 2017, 35, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Mattapally, S.; Pawlik, K.M.; Fast, V.G.; Zumaquero, E.; Lund, F.E.; Randall, T.D.; Townes, T.M.; Zhang, J. Human leukocyte antigen class I and II knockout human induced pluripotent stem cell–derived cells: Universal donor for cell therapy. J. Am. Heart Assoc. 2018, 7, e010239. [Google Scholar] [CrossRef] [Green Version]
- Flahou, C.; Morishima, T.; Takizawa, H.; Sugimoto, N. Fit-For-All iPSC-Derived Cell Therapies and Their Evaluation in Humanized Mice with NK Cell Immunity. Front. Immunol. 2021, 12, 662360. [Google Scholar] [CrossRef]
- Doss, M.X.; Sachinidis, A. Current Challenges of iPSC-Based Disease Modeling and Therapeutic Implications. Cells 2019, 8, 403. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, T.; Yasuda, S.; Sato, Y. Tumorigenicity Studies for Human Pluripotent Stem Cell-Derived Products. Biol. Pharm. Bull 2013, 36, 189–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malecki, M. “Above all, do no harm”: Safeguarding pluripotent stem cell therapy against iatrogenic tumorigenesis. Stem Cell Res. Ther. 2014, 5, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotini, A.G.; de Stanchina, E.; Themeli, M.; Sadelain, M.; Papapetrou, E.P. Escape Mutations, Ganciclovir Resistance, and Teratoma Formation in Human iPSCs Expressing an HSVtk Suicide Gene. Mol. Ther. Nucleic Acids 2016, 5, e284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traversari, C.; Marktel, S.; Magnani, Z.; Mangia, P.; Russo, V.; Ciceri, F.; Bonini, C.; Bordignon, C. The potential immunogenicity of the TK suicide gene does not prevent full clinical benefit associated with the use of TK-transduced donor lymphocytes in HSCT for hematologic malignancies. Blood 2007, 109, 4708–4715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammer, O. CD19 as an attractive target for antibody-based therapy. mAbs 2012, 4, 571–577. [Google Scholar] [CrossRef] [Green Version]
- Labenski, V.; Suerth, J.D.; Barczak, E.; Heckl, D.; Levy, C.; Bernadin, O.; Charpentier, E.; Williams, D.A.; Fehse, B.; Verhoeyen, E.; et al. Alpharetroviral self-inactivating vectors produced by a superinfection-resistant stable packaging cell line allow genetic modification of primary human T lymphocytes. Biomaterials 2016, 97, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Lim, T.T.; Geisen, C.; Hesse, M.; Fleischmann, B.K.; Zimmermann, K.; Pfeifer, A. Lentiviral Vector Mediated Thymidine Kinase Expression in Pluripotent Stem Cells Enables Removal of Tumorigenic Cells. PLoS ONE 2013, 8, e70543. [Google Scholar] [CrossRef]
- Abiko, K.; Mandai, M.; Hamanishi, J.; Matsumura, N.; Baba, T.; Horiuchi, A.; Mikami, Y.; Yoshioka, S.; Wakasa, T.; Shiozawa, T.; et al. Oct4 Expression in Immature Teratoma of the Ovary. Am. J. Surg. Pathol. 2010, 34, 1842–1848. [Google Scholar] [CrossRef]
- Villodre, E.S.; Felipe, K.B.; Oyama, M.Z.; de Oliveira, F.H.; Lopez, P.L.d.C.; Solari, C.; Sevlever, G.; Guberman, A.; Lenz, G. Silencing of the transcription factors Oct4, Sox2, Klf4, c-Myc or Nanog has different effect on teratoma growth. Biochem. Biophys. Res. Commun. 2019, 517, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.; Tang, C.; Cao, F.; Xie, X.; van der Bogt, K.; Hwang, A.; Connolly, A.J.; Robbins, R.C.; Wu, J.C. Effects of cell number on teratoma formation by human embryonic stem cells. Cell Cycle 2009, 8, 2608–2612. [Google Scholar] [CrossRef]
- Chen, T.; Wang, F.; Wu, M.; Wang, Z.Z. Development of hematopoietic stem and progenitor cells from human pluripotent stem cells. J. Cell. Biochem. 2015, 116, 1179–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindgren, A.G.; Natsuhara, K.; Tian, E.; Vincent, J.J.; Li, X.; Jiao, J.; Wu, H.; Banerjee, U.; Clark, A.T. Loss of Pten Causes Tumor Initiation Following Differentiation of Murine Pluripotent Stem Cells Due to Failed Repression of Nanog. PLoS ONE 2011, 6, e16478. [Google Scholar] [CrossRef] [PubMed]
- Herszfeld, D.; Wolvetang, E.; Langton-Bunker, E.; Chung, T.-L.; Filipczyk, A.A.; Houssami, S.; Jamshidi, P.; Koh, K.; Laslett, A.; Michalska, A.; et al. CD30 is a survival factor and a biomarker for transformed human pluripotent stem cells. Nat. Biotechnol. 2006, 24, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Sougawa, N.; Miyagawa, S.; Fukushima, S.; Kawamura, A.; Yokoyama, J.; Ito, E.; Harada, A.; Okimoto, K.; Mochizuki-Oda, N.; Saito, A.; et al. Immunologic targeting of CD30 eliminates tumourigenic human pluripotent stem cells, allowing safer clinical application of hiPSC-based cell therapy. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Martin, R.M.; Fowler, J.L.; Cromer, M.K.; Lesch, B.J.; Ponce, E.; Uchida, N.; Nishimura, T.; Porteus, M.H.; Loh, K.M. Improving the safety of human pluripotent stem cell therapies using genome-edited orthogonal safeguards. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Warlich, E.; Kuehle, J.; Cantz, T.; Brugman, M.H.; Maetzig, T.; Galla, M.; Filipczyk, A.A.; Halle, S.; Klump, H.; Schöler, H.; et al. Lentiviral vector design and imaging approaches to visualize the early stages of cellular reprogramming. Mol. Ther. 2011, 19, 782–789. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dahlke, J.; Schott, J.W.; Vollmer Barbosa, P.; Klatt, D.; Selich, A.; Lachmann, N.; Morgan, M.; Moritz, T.; Schambach, A. Efficient Genetic Safety Switches for Future Application of iPSC-Derived Cell Transplants. J. Pers. Med. 2021, 11, 565. https://doi.org/10.3390/jpm11060565
Dahlke J, Schott JW, Vollmer Barbosa P, Klatt D, Selich A, Lachmann N, Morgan M, Moritz T, Schambach A. Efficient Genetic Safety Switches for Future Application of iPSC-Derived Cell Transplants. Journal of Personalized Medicine. 2021; 11(6):565. https://doi.org/10.3390/jpm11060565
Chicago/Turabian StyleDahlke, Julia, Juliane W. Schott, Philippe Vollmer Barbosa, Denise Klatt, Anton Selich, Nico Lachmann, Michael Morgan, Thomas Moritz, and Axel Schambach. 2021. "Efficient Genetic Safety Switches for Future Application of iPSC-Derived Cell Transplants" Journal of Personalized Medicine 11, no. 6: 565. https://doi.org/10.3390/jpm11060565